СПРАВКА
Источник публикации
В данном виде документ опубликован не был.
Первоначальный текст документа также опубликован не был.
Информацию о публикации документов, создающих данную редакцию, см. в справке к этим документам.
Примечание к документу
Документ утратил силу с 01.12.2021 в части разделов "Синтетические поверхностно-активные вещества (детергенты)" и "Тяжелые металлы" (разд. 1) в связи с изданием Приказа Росгидромета от 26.07.2021 N 246. Взамен введены в действие РД 52.10.910-2021, РД 52.10.911-2021, РД 52.10.912-2021.
Документ утратил силу с 01.07.2014 в части раздела "Нефтепродукты" в связи с изданием РД 52.10.779-2013, утв. Росгидрометом 25.09.2013.
Документ утратил силу с 01.07.2014 в части раздела "Аммонийный азот" в связи с изданием РД 52.10.772-2013, утв. Росгидрометом 29.09.2013.
Документ утратил силу с 01.07.2014 в части раздела "Тяжелые металлы" (разделы 3, 4, 5) в связи с изданием РД 52.10.778-2013, утв. Росгидрометом 25.09.2013.
Документ утратил силу с 01.07.2011 в части раздела "Сероводород" в связи с изданием РД 52.10.742-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Водородный показатель (pH)" в связи с изданием РД 52.10.735-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Общий фосфор" в связи с изданием РД 52.10.739-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Кремний" в связи с изданием РД 52.10.744-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Нитриты" в связи с изданием РД 52.10.740-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Растворенный кислород в присутствии сероводорода" в связи с изданием РД 52.10.737-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Растворенный кислород" в связи с изданием РД 52.10.736-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Фосфаты" в связи с изданием РД 52.10.738-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Нитраты" в связи с изданием РД 52.10.745-2010, утв. Росгидрометом.
Документ утратил силу с 01.07.2011 в части раздела "Общая щелочность" в связи с изданием РД 52.10.743-2010, утв. Росгидрометом.
Документ введен в действие с 01.07.1993.
Название документа
"РД 52.10.243-92. Руководящий документ. Руководство по химическому анализу морских вод"
(утв. Решением Минэкологии России от 28.04.1992)
(ред. от 26.07.2021)
"РД 52.10.243-92. Руководящий документ. Руководство по химическому анализу морских вод"
(утв. Решением Минэкологии России от 28.04.1992)
(ред. от 26.07.2021)
Содержание
Решением Комитета по гидрометеорологии
и мониторингу окружающей среды
от 28 апреля 1992 года
РУКОВОДЯЩИЙ ДОКУМЕНТ
РУКОВОДСТВО ПО ХИМИЧЕСКОМУ АНАЛИЗУ МОРСКИХ ВОД
РД 52.10.243-92
Список изменяющих документов РД 52.10.745-2010, введенных в действие 1 июля 2011 года, РД 52.10.778-2013, утв. Росгидрометом 25.09.2013 РД 52.10.779-2013, утв. Росгидрометом 25.09.2013, РД 52.10.772-2013, утв. Росгидрометом 29.09.2013, Приказа Росгидромета от 26.07.2021 N 246) |
Дата введения
1 июля 1993 года
1. Утвержден Комитетом по гидрометеорологии и мониторингу окружающей среды. Решение от 28 апреля 1992 г.
2. Разработчики: С.Г. Орадовский, доктор хим. наук, профессор; Г.Г. Лятиев, канд. хим. наук; И.С. Матвеева, канд. хим. наук; Е.С. Лебедева, канд. хим. наук; А.К. Прокофьев, канд. хим. наук; И.Г. Орлова, канд. хим. наук; С.М. Черняк, канд. хим. наук; И.М. Кузнецова, В.В. Георгиевский, А.Н. Кузьмичев, В.В. Сапожников, доктор геогр. наук; Е.П. Кириллова, канд. геогр. наук, В.А. Михайлов, канд. хим. наук; Ф.А. Дмитриев, канд. хим. наук; А.В. Игнатченко, Т.В. Копылова, Т.В. Степанченко, Л.Н. Георгиевская, Е.А. Веселова
3. Согласующие организации: Управление мониторинга окружающей среды Роскомгидромета
4. Зарегистрирован в базовой организации по стандартизации и метрологии (ГОИН) за N 52.10.243-92 от 8 мая 1992 г.
5. Введен впервые
Настоящий руководящий документ (РД) распространяется на морские воды и устанавливает порядок проведения их химического анализа.
Руководство является обязательным для работников химических лабораторий Управлений по гидрометеорологии и мониторингу окружающей среды, научно-исследовательских судов и научно-исследовательских учреждений Росгидромета, других организаций Министерства экологии и природных ресурсов Российской Федерации, которые ведут гидрохимические наблюдения и исследования в морях России и Мировом океане и осуществляют мониторинг загрязнения морской среды.
"Руководство по химическому анализу морских вод" является основным методическим пособием для работников химлабораторий Управлений по гидрометеорологии и мониторингу окружающей среды, научно-исследовательских судов и научно-исследовательских учреждений Роскомгидромета, других организаций Министерства экологии и природных ресурсов Российской Федерации, проводящих гидрохимические наблюдения и исследования, мониторинг загрязнения морской среды в морях России и Мировом океане. В отличие от 1-го издания <*> настоящее Руководство полностью переработано в соответствии с требованиями Руководящего документа Госкомгидромета РД 52.24-127-87 <**>. В Руководство включены в большинстве случаев метрологически аттестованные методики химического анализа морских вод, однако сохранены и некоторые неаттестованные методики, поскольку полученные с их применением данные представляют большой научный интерес. Второе издание дополнено рядом новых методик, прежде всего касающихся определения загрязняющих веществ в морской воде: нитро-, хлор- и алкилфенолов, ксантогенатов и дитиофосфатов, гербицидов симм-триазинового ряда и группы 2,4-Д, анионных, катионных и неионогенных синтетических поверхностно-активных веществ, а также содержит аналитическую систему идентификации нефтяных разливов в море.
--------------------------------
<*> Руководство по методам химического анализа морских вод. Л.: Гидрометеоиздат, 1977.
<**> Требования к разработке, изложению, метрологической аттестации, оформлению, утверждению и внедрению методик химического анализа объектов природной среды. Методические указания. М.: Гидрометеоиздат, 1986.
В число разработчиков настоящего РД вошли составители тех разделов 1-го издания, которые не претерпели существенных изменений.
Со времени выхода в свет 1-го издания настоящего Руководства прошло 15 лет. За эти годы Государственным океанографическим институтом были разработаны и изданы еще несколько методических пособий по химическому анализу морских вод и донных отложений <*>, позволивших создать современную химико-аналитическую базу для Общегосударственной службы наблюдений за химическим состоянием морской среды (ОГСН) и научно-исследовательских учреждений гидрометеослужб России и стран СНГ, которые занимаются морскими гидрохимическими исследованиями.
--------------------------------
<*> Методические указания по определению загрязняющих веществ в морских донных отложениях, N 43. М.: Гидрометеоиздат, 1979; Методические указания по определению загрязняющих веществ в морской воде на фоновом уровне, N 45. М.: Гидрометеоиздат, 1982; Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, и др.
Новое издание Руководства призвано повысить качество и информативность морских гидрохимических данных, приблизить применяемые в нашей стране методы анализа морских вод к мировому уровню. Качество данных о химическом состоянии морской среды во многом зависит от метрологического обеспечения средств измерений и методик выполнения измерений (МВИ). Разработанные в 80-х годах Госстандартом государственные стандарты (ГОСТ), регламентирующие основные требования к МВИ, не учитывали специфику анализа проб объектов природной, в частности, морской среды, что вызвало необходимость разработки соответствующих отраслевым стандартам (ОСТ) Руководящих документов (РД) в рамках Госкомгидромета, Минводхоза, других министерств и ведомств, осуществляющих наблюдения и контроль за состоянием окружающей природной среды. Они определяли требования к метрологическим исследованиям и аттестации МВИ, что, несомненно, способствовало повышению качества получаемой информации.
При разработке настоящего Руководства соблюдались требования Руководящего документа Госкомгидромета 52.24-127-87. Вместе с тем следует подчеркнуть, что метрологические исследования и аттестация касаются только МВИ, т.е. конечной стадии химического анализа проб. Стадии же пробоотбора и пробоподготовки метрологически не исследуются и не аттестуются, так как при современном уровне метрологического обеспечения технических средств и приемов отбора, обработки, подготовки к анализу проб объектов морской и других природных сред унифицировать эти работы не представляется возможным. Между тем именно эти стадии химического анализа могут обусловливать наибольшие погрешности в результатах проведения химического мониторинга морской среды. Недостаточно корректное выполнение этих стадий анализа приводит к получению недостоверной информации, хотя затраты сил и средств на ее сбор являются, как правило, весьма значительными.
В последние годы эти проблемы оказались в центре внимания Межправительственной океанографической комиссии (МОК) ЮНЕСКО и Программы ООН по окружающей среде (ЮНЕП), которые издали ряд соответствующих руководств, рекомендованных для выполнения международных программ мониторинга загрязнения морской среды.
Разумеется, не все рекомендации этих руководств можно осуществить по чисто техническим причинам. Тем не менее при составлении отдельных разделов настоящего Руководства были учтены многие рекомендации методического пособия МОК ЮНЕСКО <**> и некоторых других документов.
--------------------------------
<**> Chemical methods for use in marine environmental monitoring. IOC Manuals and guides, N 12. UNESCO, 1983.
Для повышения информативности химического мониторинга морской среды во 2-е издание были включены новые методики определения специфических загрязняющих веществ в морской воде: нитро-, хлор- и алкилфенолов, анионных, катионных и неионогенных СПАВ, ксантогенатов и дитиофосфатов, гербицидов симм-триазинового ряда и группы 2,4-Д, общей растворенной ртути и ряда других токсичных металлов, причем все эти методики метрологически аттестованы. Были сохранены и некоторые неаттестованные методики, имеющие большое значение для информативности гидрохимических исследований океанов и морей. Метрологическая аттестация последних в настоящее время невозможна в связи с техническими трудностями. Следует также отметить, что отдельные методики, входящие в 1-е издание Руководства и не вошедшие во 2-е издание (например, колориметрические - по определению фенолов и детергентов, спектрографическая - по определению тяжелых металлов, газохроматографическая - по определению фосфорорганических пестицидов), могут применяться в практике мониторинга для полуколичественных оценок состояния загрязнения морских вод. Вместе с тем во 2-е издание включены некоторые методики (определение хлорированных углеводородов с применением капиллярной газожидкостной хроматографии, идентификация нефтяных разливов в море с применением спектрофлуорометрии, жидкостной хроматографии и капиллярной хроматографии), которые предназначены в основном для тонких гидрохимических исследований, а не для сетевых работ.
Соленостью морской воды (S+) называют выраженную в граммах суммарную массу всех твердых растворенных веществ, содержащихся в 1 кг морской воды, при условии, что все твердые вещества высушены до постоянной массы при 480 °C, органические соединения полностью минерализованы, бромиды и иодиды заменены эквивалентной массой хлоридов, а карбонаты превращены в окислы. Следовательно, морская вода в действительности содержит немного больше солей по сравнению с определенными таким образом значениями солености.
Соленость в океанографии является одной из основных характеристик водных масс, распределения морских организмов, элементов морских течений и т.д. Особую роль она играет в формировании биологической продуктивности морей и океанов, так как многие организмы очень восприимчивы к незначительным ее изменениям.
Соленость может изменяться в весьма значительных пределах, и тем не менее соотношение отдельных компонентов солевого состава морской воды остается практически постоянным, за исключением сильно опресненных районов, прилегающих к устьям рек.
Соленость морской воды определяют обычно аргентометрическим титрованием (по хлорности) и электрометрическим на солемерах [5, 6], а также комплексами "Гидрозонд" [1].
--------------------------------
<*> Настоящая методика метрологически не аттестована.
Аргентометрический метод определения солености основан на нахождении хлорности морской воды (Cl+), под которой понимают суммарную массу в граммах галогенидов (хлоридов, бромидов и иодидов), за исключением фторидов, содержащихся в 1 кг морской воды в пересчете на эквивалентное содержание хлоридов.
Хлорность определяют титрованием пробы морской воды раствором нитрата серебра AgNO3 до полного осаждения всех галогенидов, кроме фторидов.
Количественно значение хлорности определяют из соотношения:
Cl = 0,3285234Ag,
Ag - масса химически чистого серебра в граммах, необходимая для осаждения всех галогенидов, содержащихся в 1 кг морской воды, а значение солености - из соотношения:
S = 1,80655Cl.
Значения солености, вычисляемые в интервале 32 - 38 + по старому уравнению Кнудсена S = 0,030 + 1,8050Cl и по новой формуле, отличаются не более чем на 0,0026+.
Пробу морской воды титруют раствором нитрата серебра, используя в качестве индикатора хромат калия. Поскольку массу солей выражают в граммах, а массу воды - в килограммах, то в результаты объемного химического анализа необходимо вводить поправку на плотность пробы. Для этого в качестве стандарта применяют так называемую нормальную воду, с помощью которой устанавливают титр раствора нитрата серебра. Определив аргентометрическим методом хлорность пробы морской воды, находят затем по таблицам [3] ее соленость, выраженную непосредственно в единицах массы (промилле).
Важно отметить, что этим методом можно определять соленость только таких морских вод, для которых выпускается нормальная вода, т.е. вод открытых частей морей и океанов. Если же массы пробы и нормальной воды отличаются, то в результаты титрования вводят поправки к отсчетам по бюретке. При определении хлорности солоноватых вод Каспийского, Азовского и Аральского морей необходимо использовать таблицы [4].
В случае же сильно опресненных вод (S <= 1+) настоящий метод вообще нельзя применять [5].
Для выполнения анализа применяются:
мешалка магнитная по ТУ 25-11-834;
бюретки морские автоматические калиброванные на 15 мл (рис. 1);

Рис. 1. Автоматические бюретки и пипетки ГОИН разных типов
а - бюретка для определения хлорности морской воды с высокой
соленостью (крупно дано устройство для автоматической
установки нулевого деления бюретки) с раствором
азотнокислого серебра; б - бюретка для определения солености
в широком диапазоне; в - бюретка для определения
низкой солености; г - пипетка.
Кнудсена - для всех соленостей,
ГОИН - для высокой солености,
ГОИН - для всех соленостей,
ГОИН - для низкой солености;
пипетка автоматическая на 15 мл по ГОСТ 20292;
толстостенный химический стакан на 100 - 150 мл по ГОСТ 25336;
склянка с притертой пробкой и колпаком на 300 мл для хранения нормальной воды по ТУ 6-19-6;
капельница для индикатора по ТУ 25-11-1126;
бутыль из темного стекла (или из светлого, но покрытого снаружи сплошным слоем черной краски) на 5 - 10 л для хранения азотнокислого серебра по ТУ 6-19-45;
промывалка на 0,5 - 1,0 л для дистиллированной воды по ТУ 64-1-596;
палочка стеклянная для перемешивания титруемой пробы (при отсутствии магнитной мешалки) по ТУ 25-11-1049;
банка (склянка) с широким горлом для сливания остатков хлористого серебра по ТУ 6-19-6;
нормальная морская вода соленостью 35+;
серебро азотнокислое, х.ч., по ГОСТ 1277;
калий хромовокислый, х.ч., по ГОСТ 4459.
Отбор проб морской воды для определения солености производят после взятия проб на pH и растворенный кислород. Пробы отбирают из батометра через резиновую трубку в любые склянки объемом 100 - 250 мл с хорошо подогнанными резиновыми пробками. Перед взятием пробы склянки 2 - 3 раза ополаскивают водой из батометра и затем заполняют водой, но не до пробок во избежание выталкивания при изменении температуры. В случае длительного хранения проб необходимо надеть поверх пробок резиновые колпачки.
Допускается хранение проб в течение нескольких недель в склянках, закрытых восковыми пробками (их отмачивают 30 - 40 с в расплавленном парафине, дают стечь его избытку и высушивают на доске на воздухе), а также в целиком запарафинированных склянках. В таком виде пробы хранятся несколько лет без изменения солености [5].
Определение солености при вскрытии склянок нельзя задерживать более чем на час.
1. Нормальная морская вода служит основным стандартным раствором. Она представляет собой фильтрованную океаническую воду, хлорность которой близка к 19,38+, что соответствует солености 35,00+, т.е. средней солености воды океана, поэтому она и называется нормальной. Этот стандартный раствор со значением хлорности, определенным до третьего знака после запятой включительно, поступает в лаборатории в запаянных стеклянных баллонах емкостью 250 мл <*>. Перед работой трубочки баллона надрезают напильником и отламывают, а нормальную воду переливают в чистую склянку с пришлифованной пробкой и колпаком.
--------------------------------
<*> В СНГ нормальную воду изготавливает аналитическая лаборатория Института океанологии РАН.
2. Раствор азотнокислого серебра готовят растворением 37,1 г нитрата серебра в дистиллированной воде в мерной колбе на 1 л. Обычно приготовляют 5 - 10 л раствора и хранят его в темной бутыли. Раствор должен быть совершенно прозрачным. Если же он мутный, то его отстаивают в темном месте до полного просветления и затем сифонируют в чистую бутыль.
3. Раствор индикатора - хромовокислого калия получают растворением 10 г чистой соли в 90 мл дистиллированной воды (10%-ный раствор).
1.4.2. Определение поправки к титру раствора азотнокислого серебра по нормальной воде
Ополоснув предварительно пипетку нормальной водой, переносят 15,0 мл ее в стакан для титрования и после добавления пяти капель индикатора титруют раствором азотнокислого серебра. Во время титрования раствор должен энергично перемешиваться. До появления оранжевых пятен труднорастворимого оранжевого хромата серебра раствор титруют при полностью открытом кране, а затем по каплям. Титрование заканчивают после появления слабой оранжевой окраски осадка, не исчезающей при перемешивании в течение 20 с. Через 15 с записывают отсчет бюретки с точностью до 0,01 деления. Затем титрование проводят вторично при строгом соблюдении тех же условий. Расхождение в отсчетах двух последовательных титрований не должно превышать 0,01 деления, в противном случае выполняют третье титрование. Если же и в этом случае расхождение превышает указанное значение, то необходимо еще раз тщательно перемешать раствор азотнокислого серебра в бутыли и обратить внимание на единообразие в процессе титрования. При вычислении поправки берут среднее арифметическое результатов двух последовательных титрований.
Разность  между отсчетом бюретки (A) и хлорностью нормальной воды (N), указанной на этикетке баллона, не должна выходить за пределы +0,145 или -0,150 делений. Если
между отсчетом бюретки (A) и хлорностью нормальной воды (N), указанной на этикетке баллона, не должна выходить за пределы +0,145 или -0,150 делений. Если  выходит за эти пределы, то приготовленный раствор либо крепче (A < N), либо слабее (A > N), чем должен быть, и поэтому необходимо добавить соответствующее количество дистиллированной воды или нитрата серебра.
выходит за эти пределы, то приготовленный раствор либо крепче (A < N), либо слабее (A > N), чем должен быть, и поэтому необходимо добавить соответствующее количество дистиллированной воды или нитрата серебра.
Расчет исправления концентрации раствора азотнокислого серебра проводят по следующим формулам:
1) раствор крепче нормы, т.е.  , тогда:
, тогда:
, тогда: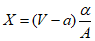 ,
,где: Х - объем дистиллированной воды, который необходимо добавить в раствор, мл; V - первоначальный объем раствора, мл; a - объем раствора, израсходованного на промывку бюретки, мл; A - объем раствора, пошедшего на титрование нормальной воды, мл; N - хлорность нормальной воды;  - абсолютное значение разности N - A;
- абсолютное значение разности N - A;
2) раствор слабее нормы, т.е. A > N (N - A =  ), тогда:
), тогда:
 ,
,где X - масса азотнокислого серебра, которое необходимо добавить в раствор, г.
Для быстрого нахождения количества дистиллированной воды или азотнокислого серебра, которое необходимо добавить в раствор для приведения величины  в пределы, допускаемые "Океанографическими таблицами" [3], очень удобна номограмма, изображенная на рис. 2.
в пределы, допускаемые "Океанографическими таблицами" [3], очень удобна номограмма, изображенная на рис. 2.
в пределы, допускаемые "Океанографическими таблицами" [3], очень удобна номограмма, изображенная на рис. 2.
Рис. 2. Номограмма для приведения концентрации раствора
азотнокислого серебва к нормальному значению
На номограмме горизонтальные линии отвечают определенным значениям  , причем в правом столбце
, причем в правом столбце 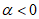 , а в левом
, а в левом  . Отыскав на диагонали точку, соответствующую определенному значению
. Отыскав на диагонали точку, соответствующую определенному значению  , и проведя от этой точки вертикальную линию вниз, когда
, и проведя от этой точки вертикальную линию вниз, когда 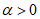 , или вверх, когда
, или вверх, когда  , на нижней горизонтали находят объем воды, мл, а на верхней - массу азотнокислого серебра, г, которую необходимо добавить в расчете на каждый литр раствора азотнокислого серебра. После изменения концентрации раствора необходимо вновь определить его титр по нормальной воде.
, на нижней горизонтали находят объем воды, мл, а на верхней - массу азотнокислого серебра, г, которую необходимо добавить в расчете на каждый литр раствора азотнокислого серебра. После изменения концентрации раствора необходимо вновь определить его титр по нормальной воде.
, причем в правом столбце К титрованию приступают только тогда, когда температура проб морской воды достигнет комнатной. Для этого их необходимо выдержать в помещении лаборатории не менее часа.
Сполоснув исследуемой водой пипетку, отбирают 15,0 мл пробы и переносят ее в химический стакан. Титрование проводят аналогично определению поправки раствора азотнокислого серебра по нормальной воде. Контрольные титрования нормальной воды следует проводить при изменении условий освещения или температуры воздуха, а также после 15 - 20 титрований проб.
Оттитрованную пробу с осадком хлористого серебра сливают в склянку для остатков серебра. По ее заполнении отстоявшуюся жидкость декантируют и выбрасывают, а хлористое серебро собирают, высушивают и сдают для регенерации.
Стакан для титрования не обязательно ополаскивать дистиллированной водой от частиц хлористого серебра при правильном титровании, однако если проба перетитрована, то перед внесением в стакан следующей пробы его следует тщательно промыть.
При возникновении каких-либо сомнений в правильности титрования необходимо его повторить.
По окончании работы пипетку заполняют дистиллированной водой, а бюретку - раствором азотнокислого серебра и накрывают последнюю чехлом из плотной черной материи.
После окончания титрования проб вычисляют хлорность по формуле
Cl = a + k,
где: a - исправленный отсчет бюретки после титрования пробы; k - поправка титрования, определяемая по "Океанографическим таблицам" [3].
Чтобы найти по таблицам k, необходимо вычислить значение 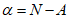 (разность между хлорностью нормальной воды N, по которой определялся титр раствора азотнокислого серебра, и исправленным отчетом бюретки A после титрования нормальной воды). В графе
(разность между хлорностью нормальной воды N, по которой определялся титр раствора азотнокислого серебра, и исправленным отчетом бюретки A после титрования нормальной воды). В графе  таблиц [3] находят соответствующее значение и, согласно отсчету по бюретке a определяют значение k. Далее в табл. 1.5 "Соотношение значений величин Cl+, S+,
таблиц [3] находят соответствующее значение и, согласно отсчету по бюретке a определяют значение k. Далее в табл. 1.5 "Соотношение значений величин Cl+, S+,  ,
, 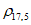 " находят значение S+.
" находят значение S+.
(разность между хлорностью нормальной воды N, по которой определялся титр раствора азотнокислого серебра, и исправленным отчетом бюретки A после титрования нормальной воды). В графе Необходимо иметь в виду, что таблицы соотношения значений хлорности, солености и условной плотности  рассчитаны для океанической воды, имеющей строго определенные соотношения солеобразующих ионов, и поэтому ими можно пользоваться только для морей, имеющих хороший водообмен с океаном. Для морей, изолированных от океана и имеющих затрудненный водообмен с ним (Каспийское, Аральское) или подверженных сильному воздействию берегового стока (Азовское море), пользоваться "Океанографическими таблицами" [3] нельзя вследствие отличия солевого состава вод этих морей от вод океана. В этом случае надо пользоваться справочными таблицами [4].
рассчитаны для океанической воды, имеющей строго определенные соотношения солеобразующих ионов, и поэтому ими можно пользоваться только для морей, имеющих хороший водообмен с океаном. Для морей, изолированных от океана и имеющих затрудненный водообмен с ним (Каспийское, Аральское) или подверженных сильному воздействию берегового стока (Азовское море), пользоваться "Океанографическими таблицами" [3] нельзя вследствие отличия солевого состава вод этих морей от вод океана. В этом случае надо пользоваться справочными таблицами [4].
Электрометрический метод определения солености основан на измерении относительной электропроводности морской воды с помощью бесконтактного индукционного солемера, что позволяет ускорить и в принципе увеличить точность ее определения по сравнению с аргентометрическим титрованием.
В СНГ в судовых условиях используется солемер ГМ-65, принцип работы которого основан на изменении электропроводности морской воды в зависимости от количества растворенных в ней солей. Измерения проводят бесконтактным датчиком, состоящим из двух индуктивно связанных трансформаторов с автоматической компенсацией влияния температуры на электропроводность.
Значения электропроводности в относительных единицах с помощью "Международных океанологических таблиц" [2] переводят в соленость. Принципиальная схема прибора приведена на рис. 3.

Рис. 3. Упрощенная схема солемера
TP1, TP2 - тороидальные трансформаторы; Wд - обмотка
генератора; Wд - обмотка делителя; Wк - компенсационная
обмотка; Rв - сопротивление короткозамкнутого жидкостного
витка; R1, R2, Rt - сопротивление цепи термокомпенсации.
Пробу морской воды заливают в датчик - прозрачный сосуд, в котором жестко укреплены тороидальные трансформаторы, T1 и T2,являющиеся индикаторами электропроводности. Они индуктивно связаны друг с другом короткозамкнутым жидкостным витком с сопротивлением Rв, причем сила этой связи зависит от внутреннего объема датчика, геометрических размеров тороидов и электропроводности воды. Трансформаторы также связаны компенсирующей цепью из обмотки делителя Wg, компенсационной обмотки Wк и цепи термокомпенсации R1, R2, Rt. Поскольку Wв и Wк имеют противоположные полярности обмоток, то магнитные потоки, создаваемые в сердечнике T2 токами Iв и Iк, направлены друг к другу.
Генератор низкой частоты питает первичную обмотку T1, а на вторичной обмотке можно добиться отсутствия сигнала на входе усилителя при регулировании напряжения путем изменения числа витков Wg. Электрическая проводимость прямо пропорциональна числу витков, что позволяет проводить калибровку в единицах электропроводности, которую измеряют по показаниям делителя напряжения декадных трансформаторов в относительных единицах.
Для устранения влияния температуры на результаты измерения электропроводности морской воды в солемере предусмотрена термокомпенсация с помощью термистора Rt, соединенного последовательно с обмоткой Wк. Компенсацию в рабочем диапазоне значений температуры производят переменным шунтирующим сопротивлением R2, которое устанавливают при изменении температуры в помещении на 1 - 2 °C. К каждому прибору прилагается таблица зависимости величины R2 от температуры. Погрешность 0,01 °C вследствие неполной термокомпенсации вносит в определяемое значение солености ошибку 0,01+.
Солемер ГМ-65 используют как в стационарных условиях, так и на борту судна при температуре воздуха и проб от 10 до 35 °C, относительной влажности до 98% при 20 °C и атмосферном давлении в диапазоне 9600 - 10400 Н/м2.
Необходимо отметить, что электропроводность изменяется в зависимости от давления не более чем на 2,5 - 10% начиная с глубины 500 м [1].
Для выполнения анализа применяются:
солемер ГМ-65;
склянки для нормальной и субнормальной воды емкостью 300 мл с пришлифованными пробками и колпаками (кислотные склянки) по ТУ 6-19-6;
нормальная вода (см. п. 1.4.1).
Пробы морской воды отбираются так же, как указано в п. 1.3.
2.4.1. Солемер вместе с посудой с пробами морской воды и нормальной водой необходимо установить в точке, защищенной от прямого попадания солнечных лучей, теплового влияния приборов и воздействия конвективных потоков воздуха с резкими колебаниями температуры. Температура в помещении не должна изменяться более чем на 1 - 2 °C. Приступать к измерениям можно только после выравнивания температуры помещения, прибора, проб морской и нормальной вод.
2.4.2. Нормальную воду из запаянных баллонов переливают в кислотные склянки. Очень удобно для текущей работы с целью уменьшения влияния тепловой инерции морской и нормальной вод на скорость измерений солености отбирать их в систему, состоящую из двух склянок (рис. 4).

Рис. 4. Рекомендуемый способ хранения проб морской воды,
эталонного раствора нормальной воды и дистиллированной воды,
для текущей работы на приборе
1 - колба; 2, 3 - пробки резиновые; 4 - трубка стеклянная;
5 - трубка резиновая; 6 - палочка стеклянная;
7 - трубка стеклянная; 8 - промывалка
Воду засасывают в датчик (ячейку) солемера через пропущенную сквозь резиновую пробку 3 стеклянную трубку 4 диаметром около 10 мм, доходящую до дна колбы 1, объемом 800 - 1000 мл. На верхний конец трубки 4 надевают кусок резиновой трубки 5, закрытой стеклянной палочкой 6. Давление в колбе регулируют стеклянной трубкой 7, проходящей сквозь пробку 3 и соединенной с небольшой промывалкой 8, в которой находится такая же вода, что и в колбе, тем самым предотвращая испарение. Эта система особенно удобна в тропических условиях.
2.4.3. Датчик наполняют пробой следующим образом. Надевают левый шланг датчика на трубку 4, после чего открывают его левый кран и закрывают правый. Поворотом ручки насоса наполняют измерительную камеру до появления пробы в сливной камере. Последнюю нельзя заполнять целиком, так как при этом выходит из строя насос. Если хода насоса не хватает для наполнения датчика, то следует закрыть левый и открыть правый краны, вернуть ручку насоса в исходное положение, затем закрыть правый и открыть левый краны и продолжить заполнение датчика пробой. При появлении пробы в сливной камере закрывают левый и открывают правый краны, и излишки пробы сливают.
При заполнении датчика пробой выключатель "питание" должен быть выключен.
Для предотвращения появления пены и пузырьков воздуха в датчике необходимо отрегулировать скорость вращения мешалки путем поворота оси переменного сопротивления через отверстие в кране датчика, а также быстроту наполнения последнего путем изменения скорости угла поворота ручки насоса.
Необходимо помнить, что присутствие пузырьков воздуха в измерительной камере абсолютно недопустимо.
2.4.4. При измерении солености на солемере расходуется много нормальной воды. Поэтому допускается использование субнормальной воды, которая представляет собой морскую воду известной хлорности, приготовленную самостоятельно в лаборатории или на судне и проверенную по нормальной воде. Для ее приготовления берут морскую воду (отбор делать в открытом океане ниже глубины 50 м) с хлорностью выше 18+. Пробу стабилизируют добавлением нескольких кристаллов тимола и затем быстро переливают в бутыль.
Ее хлорность определяют титрованием относительно нормальной воды, причем расхождение двух последовательных определений не должно превышать 0,02 делений бюретки. В этом случае берут среднее из двух определений.
Субнормальную воду хранят в темной бутыли, плотно закрытой запарафинированной пробкой с сифонной трубкой, через которую производят ее отбор. Хлорность субнормальной воды необходимо проверять не реже одного раза в неделю, причем она не должна изменяться более чем на 0,02+.
2.4.5. После подготовки рабочего места к работе проверяют солемер. Для этого поворотом арретира ставят при отключенном приборе стрелку индикатора на нуль, подключают шнур питания к батарее аккумуляторов, включают тумблеры "питание" и "нагрев", переключатель " - k - t" переводят в положение "
- k - t" переводят в положение " ". Стрелка индикатора должна находиться на окрашенном участке шкалы, в противном случае необходимо зарядить аккумуляторы. При отсутствии реакции стрелки индикатора необходимо поменять его полярность.
". Стрелка индикатора должна находиться на окрашенном участке шкалы, в противном случае необходимо зарядить аккумуляторы. При отсутствии реакции стрелки индикатора необходимо поменять его полярность.
2.4.6. Температуру нормальной воды и проб морской воды измеряют ртутным термометром с точностью +/- 0,2 °C, причем разность температур не должна превышать +/- 0,5 °C. По таблице приложения 1 к паспорту прибора определяют положение переключателя "компенсация", соответствующее измеренной температуре.
2.4.7. Калибровка солемера
Калибровка солемера производится в соответствии с инструкцией к прибору.
2.4.8. Проверка температурной компенсации
Необходимо не реже одного раза в шесть месяцев производить проверку температурной компенсации солемера. Для этого его калибруют (см. выше), после чего включают на 3 - 5 мин тумблер "нагрев" и через 3 мин после его выключения измеряют относительную электропроводность. Разность между начальным и измеренным значениями электропроводности не должна превышать +/- 0,0002 при изменении температуры в пределах, указанных в таблице приложения 1 к паспорту солемера. В этом случае компенсацию можно считать удовлетворительной. Затем проводят проверку точности компенсации во всем рабочем диапазоне температур, т.е. от 10 до 35 °C. Если же относительная электропроводность изменяется более чем на +/- 0,0002, то таблицу приложения 1 надо составить заново.
Для этого 1 - 2 л нормальной воды выдерживают полчаса при 50 °C для удаления растворенных в ней газов и переливают в закрытую литровую колбу. Относительную электропроводность начинают измерять при 10 °C. Температуру пробы изменяют переключателем "нагрев" и измеряют температуру через 2 - 3 мин после его включения. Экспериментально находят такие значения положений переключателя "компенсация" в рабочем диапазоне температур, при которых обеспечивается температурная компенсация по относительной электропроводности не менее чем на +/- 0,0002. Если относительная электропроводность возрастает при нагреве воды на 1 - 2 °C, то следует увеличить значение "компенсации", если убывает - уменьшить. Полученные данные записывают в таблицу приложения 1 к прибору.
Склянки с пробами выдерживают 2 - 3 ч недалеко от солемера для принятия ими температуры лабораторного помещения. Измерения электропроводности проб морской воды следует проводить в порядке возрастания солености, поскольку в этом случае сокращается число промывок и, следовательно, повышается производительность работы на солемере.
После калибровки солемера из датчика сливают нормальную воду и промывают его пробой один-два раза. Затем вновь заполняют ячейку пробой, причем появляющиеся пузырьки воздуха следует удалить. Проверяют температуру пробы, которая должна находиться в пределах установленной термокомпенсации.
Калибровка прибора сохраняется для всей серии проб. Значение электропроводности отсчитывают по показаниям лимбов переключателя "электропроводность", при этом стрелка индикатора должна быть установлена на нуль. Затем пробу сливают и в датчик наливают новую.
Промывать ячейку пробой каждый раз не обязательно, однако это необходимо делать при проверке калибровки, при скачке солености и при измерении первой пробы следующей станции.
При обнаружении разброса показаний прибора необходимо делать повторные калибровки солемера по нормальной или субнормальной воде через каждые 10 - 15 проб. Если прибор работает стабильно, то его проверку можно производить реже, через 20 - 30 проб.
По окончании анализа всей серии проб проверяют калибровку солемера, после чего ячейку несколько раз промывают дистиллированной водой.
Полученные значения электропроводности проб морской воды переводят в соленость по "Международным океанологическим таблицам" [2]. Необходимо отметить, что эти таблицы нельзя использовать для распресненных морских вод.
Определение электропроводности и расчет солености распресненных морских вод можно проводить согласно "Методическим указаниям" <*>. Следует, однако, подчеркнуть, что погрешности этих определений метрологически не установлены.
--------------------------------
<*> См. Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, с. 6 - 13.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 1 по 31 октября 1986 г. (табл. 1), настоящая методика определения солености морских вод допущена к применению в организациях Росгидромета.
Таблица 1
Результаты метрологической аттестации
Диапазон изменения солености, + | Показатель воспроизводимости ( | Показатель правильности ( | Показатель погрешности МВИ, суммарная погрешность ( |
33,9 - 35,1 | 0,0068 | 0,030 | 0,030 |
--------------------------------
<*> Настоящая методика метрологически не аттестована.
В п. 1 уже говорилось, что аргентометрическое определение хлорности с последующим вычислением солености по "Океанографическим таблицам" [3] возможно только для вод открытых морей и океанов, для которых существует нормальная вода, т.е. имеет место строгое соответствие хлорности, солености и плотности морской воды. Однако это важнейшее условие заметно нарушается на приустьевых взморьях больших рек вследствие сильного разбавления морских вод речными водами, имеющими другой солевой состав и, что особенно важно, гораздо более низкую концентрацию хлорид-иона.
Вместе с тем определение концентрации хлорид-иона в опресненных водах имеет немаловажное практическое значение для объяснения химических процессов, происходящих в море. Для расчетов солености распресненных вод часто используют "хлорные коэффициенты" - отношение содержания какого-либо компонента морской воды к хлорид-иону (например,  - сульфатно-хлорный коэффициент). Поэтому для распресненной морской воды определение хлорид-иона является обязательным.
- сульфатно-хлорный коэффициент). Поэтому для распресненной морской воды определение хлорид-иона является обязательным.
- сульфатно-хлорный коэффициент). Поэтому для распресненной морской воды определение хлорид-иона является обязательным.Распресненной морской водой принято считать воду, в которой содержится до 1+ хлорид-иона. Естественно, что концентрация других солей будет незначительна, что приближает ее плотность к пресной воде. Поэтому содержание хлорид-иона в сильно опресненной воде удобнее относить к 1 л, а не к 1 кг, как это принято для собственно морской воды, и количественно выражать в мг/л.
В распресненных морских водах хлорность определяют так же, как и в собственно морских водах, т.е. аргентометрическим титрованием, но с применением более низких концентраций рабочих растворов.
Для выполнения анализа применяются:
мешалка магнитная по ТУ 25-11-834;
бюретка автоматическая калиброванная на 50 мл;
пипетки автоматические калиброванные на 100; 50; 25; 5 и 1 мл по ГОСТ 20292;
колбы мерные на 1000; 200 и 100 мл по ГОСТ 1770;
колба коническая на 250 мл по ГОСТ 25336;
капельница для индикатора по ТУ 25-11-1126;
бутыль из темного стекла на 2 - 3 л для хранения азотнокислого серебра по ТУ 6-19-45;
промывалка на 0,5 - 1,0 л для дистиллированной воды по ТУ 64-1-596;
палочка стеклянная для перемешивания титруемой пробы (при отсутствии магнитной мешалки) по ТУ 25-11-1049;
банка (склянка) с широким горлом для сливания остатков хлористого серебра по ТУ 6-19-6;
серебро азотнокислое, х.ч., по ГОСТ 1277;
натрий хлористый, х.ч., по ГОСТ 4233;
калий хромовокислый, х.ч., по ГОСТ 4459.
Отбор и хранение проб распресненной морской воды производят аналогично морским водам нормальной солености, за исключением того, что объем отбираемой воды должен быть не менее 200 - 250 мл.
3.4.1. Методы приготовления реактивов для проведения анализа
1. Растворы азотнокислого серебра для получения необходимой точности определения хлорности готовят двух концентраций хлорид-иона - 2,5 и 1 мг/мл.
Их готовят растворением соответственно 12,0 и 4,8 г нитрата серебра в дистиллированной воде в мерной колбе на 1 л и хранят в темных бутылях. Второй раствор можно также получить при разведении 400 мл первого раствора дистиллированной водой в мерной колбе на 1 л. Однако этот способ менее точен и им можно пользоваться лишь в исключительных случаях.
2. Стандартные растворы хлористого натрия используют для установки титра рабочих растворов нитрата серебра. Для этого хлористый натрий прокаливают в фарфоровой чашке при 500 - 600 °C в электропечи или на горелке при постоянном помешивании стеклянной палочкой до прекращения характерного потрескивания соли. Ее хранят в бюксе в эксикаторе над хлористым кальцием. Готовят два раствора хлористого натрия концентрациями 2,5 и 1,0 мг/мл. Их готовят растворением 4,1210 и 1,6884 г соответственно хлористого натрия в дистиллированной воде в мерной колбе на один литр. Для работы на борту судна эти навески необходимо готовить заблаговременно в береговой лаборатории и хранить их до употребления в хорошо пришлифованных колбочках или бюксах, а лучше всего в запаянных ампулах.
3. Раствор индикатора готовят растворением 10 г химически чистого хромата калия в 90 мл дистиллированной воды (10%-ный раствор).
3.4.2. Определение титра раствора азотнокислого серебра
Перед началом титрования проб воды необходимо проверить титр каждого из полученных растворов нитрата серебра с применением стандартных растворов хлористого натрия с точными титрами хлорид-иона 2,5 и 1,0 мг/мл соответственно. Для этого калиброванную пипетку трижды ополаскивают небольшим количеством используемого раствора хлористого натрия и переносят ею в коническую колбу 25 мл этого раствора, после чего туда же добавляют 75 мл дистиллированной воды из мерного цилиндра. В полученные 100 мл раствора прибавляют 1 мл раствора хромата калия и при энергичном перемешивании титруют соответствующим раствором нитрата серебра. Конец реакции определяют по появлению слабой оранжевой окраски осадка (аналогично титрованию нормальной воды). Титрование проводят дважды и берут средний результат. Титр раствора нитрата серебра вычисляют по формуле
 ,
,где a - исправленный объем пипетки; c - истинное содержание 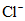 в 1 мл раствора хлористого натрия; n - исправленный объем бюретки после титрования пробы.
в 1 мл раствора хлористого натрия; n - исправленный объем бюретки после титрования пробы.
Значение  записывают с точностью до 0,001 мг. Титр раствора азотнокислого серебра при больших сериях проб проверяют как до титрования, так и после него в конце вахты или рабочего дня.
записывают с точностью до 0,001 мг. Титр раствора азотнокислого серебра при больших сериях проб проверяют как до титрования, так и после него в конце вахты или рабочего дня.
Пробы переносят в помещение лаборатории на 2 - 3 ч для выравнивания температуры.
Перед их титрованием необходимо выбрать концентрацию раствора нитрата серебра. Для этого в маленькую коническую колбочку отмеривают 5 мл пробы, прибавляют две капли индикатора и титруют раствором нитрата серебра, 1 мл которого содержит около 2,5 мг  . Если на титрование пошло более 2; 1 - 2 или менее 1 мл, то содержание
. Если на титрование пошло более 2; 1 - 2 или менее 1 мл, то содержание  составляет соответственно более 1000; 500 - 1000 и менее 500 мг/л.
составляет соответственно более 1000; 500 - 1000 и менее 500 мг/л.
По результатам предварительных титрований определяют способ титрования. В первом случае используют тот же метод, что и при титровании проб нормальной солености, во втором - титруют первым раствором (1 мл нитрата серебра содержит 2,5 мг  ), в третьем - вторым раствором (1 мл нитрата серебра содержит 1 мг
), в третьем - вторым раствором (1 мл нитрата серебра содержит 1 мг  ).
).
После нахождения нужной концентрации рабочего раствора приступают к титрованию проб. При концентрации  более 500 мг/л отмеривают в коническую колбу калиброванной пипеткой 50 мл пробы, а при концентрациях
более 500 мг/л отмеривают в коническую колбу калиброванной пипеткой 50 мл пробы, а при концентрациях 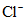 менее 500 мг/л берут 100 мл пробы. После добавления 1 мл индикатора пробу титруют при энергичном перемешивании до появления слабой оранжевой окраски осадка, не исчезающей в течение 20 с после добавления последней капли нитрата серебра. Через 30 с после окончания титрования записывают в журнал отсчет бюретки. Оттитрованную пробу сливают в банку (склянку) для хранения отходов серебра.
менее 500 мг/л берут 100 мл пробы. После добавления 1 мл индикатора пробу титруют при энергичном перемешивании до появления слабой оранжевой окраски осадка, не исчезающей в течение 20 с после добавления последней капли нитрата серебра. Через 30 с после окончания титрования записывают в журнал отсчет бюретки. Оттитрованную пробу сливают в банку (склянку) для хранения отходов серебра.
Результаты титрования вычисляют по формуле
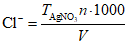 ,
,где n - исправленный объем бюретки, мл; 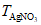 - точный титр раствора AgNO3, мг/мл; V - исправленный объем титрованной пробы, мл.
- точный титр раствора AgNO3, мг/мл; V - исправленный объем титрованной пробы, мл.
Определение солености и хлорности может выполнять инженер или техник-химик со средним специальным образованием, имеющий опыт работы с химическими препаратами.
5.1. Для анализа солености воды в 10 пробах аргентометрическим методом требуется 2,6 чел.-ч, в том числе:
на взятие проб из батометра - 0,2 чел.-ч;
на приготовление растворов реактивов - 0,6 чел.-ч;
на подготовку посуды - 0,3 чел.-ч;
на фильтрование раствора азотнокислого серебра - 0,3 чел.-ч;
на выполнение измерений - 0,7 чел.-ч;
на выполнение расчетов - 0,5 чел.-ч.
5.2. Для анализа солености воды в 10 пробах электрометрическим методом требуется 1,6 чел.-ч, в том числе:
на взятие проб из батометра - 0,2 чел.-ч;
на подготовку посуды - 0,4 чел.-ч;
на калибровку солемера - 0,3 чел.-ч;
на выполнение измерений - 0,3 чел.-ч;
на выполнение расчетов 0,4 чел.-ч.
5.3. Для анализа хлорности воды в 10 пробах требуется 3,1 чел.-ч, в том числе:
на взятие проб из батометра - 0,2 чел.-ч;
на подготовку реактивов - 1,0 чел.-ч;
на подготовку посуды - 0,4 чел.-ч;
на выполнение измерений - 0,9 чел.-ч;
на выполнение расчетов - 0,6 чел.-ч.
1. Временные методические указания для работ с измерительным комплексом "Зонд-батометр". Одесса: изд. ОдО ГОИН, 1976. 70 с.
6. Федосов М.В., Орадовский С.Г. Определение солености морской воды. В кн.: Современные методы рыбохозяйственных морских гидрохимических исследований. М.: Пищевая промышленность, 1973, с. 37 - 44.
Раздел исключен с 1 июля 2011 года. - РД 52.10.743-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.735-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.736-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.737-2010, утв. Росгидрометом.
Сероводород и сернистые соединения, сульфиды и другие восстановленные формы серы не являются типичными и постоянными компонентами морских вод.
Однако при определенных условиях сероводород и сульфиды могут накапливаться в глубоких слоях моря в значительных количествах. Области с достаточно высоким содержанием сероводорода могут временами образовываться даже на небольших глубинах. Но и временное накопление сероводорода в море нежелательно, так как его появление вызывает гибель морской фауны. Вместе с тем присутствие сероводорода в морской воде служит характерным показателем определенных гидрологических условий, а также интенсивного потребления растворенного кислорода и наличия большого количества легко окисляющихся веществ различного происхождения.
Основным источником возникновения сероводорода в море служит биохимическое восстановление растворенных сульфатов (процесс десульфатации). Десульфатация в море вызывается жизнедеятельностью особого вида анаэробных десульфатирующих бактерий, которые восстанавливают сульфаты в сульфиды, последние же разлагаются растворенной угольной кислотой до сероводорода.
Схематически этот процесс можно представить следующим образом:
 ,
, .
.В действительности указанный процесс протекает более сложно, и в сероводородной зоне присутствует не только свободный сероводород, но и другие формы продуктов восстановления сульфатов (сульфиды, гидросульфиты, гипосульфиты и др.).
В гидрохимической практике содержание восстановленных форм соединений серы принято выражать в эквиваленте сероводорода. Лишь в особых специально поставленных исследованиях различные восстановленные формы серы определяются раздельно. Эти определения здесь не рассматриваются.
Вторым источником возникновения сероводорода в море служит анаэробный распад богатых серой белковых органических остатков отмерших организмов. Содержащие серу белки, распадаясь в присутствии достаточного количества растворенного кислорода, окисляются, и содержащаяся в них сера переходит в сульфат-ион. В анаэробных условиях распад серосодержащих белковых веществ ведет к образованию минеральных форм серы, т.е. сероводорода и сульфидов.
Случаи временного возникновения анаэробных условий и связанного с ними накопления сероводорода наблюдаются в Балтийском и Азовском морях, а также в некоторых губах и заливах других морей.
Классическим примером морского бассейна, зараженного сероводородом, является Черное море, где лишь верхний сравнительно тонкий поверхностный слой свободен от сероводорода.
Возникающие в анаэробных условиях сероводород и сульфиды легко окисляются при поступлении растворенного кислорода, например при ветровом перемешивании верхних, хорошо аэрированных слоев воды с глубинными водами, зараженными сероводородом.
Поскольку даже временное накопление сероводорода и сернистых соединений в море имеет существенное значение как показатель загрязнения вод и возможности возникновения заморов морской фауны, наблюдения за его появлением совершенно необходимы при изучении гидрохимического режима моря.
--------------------------------
<*> Настоящая методика метрологически не аттестована.
Сероводород присутствует в морской воде в виде растворенной, слабодиссоциированной сероводородной кислоты H2S, а также в виде гидросульфидного  и сульфидного
и сульфидного  ионов.
ионов.
До настоящего времени вопрос о равновесии системы сероводорода и о формах сероводорода в морской воде исследован далеко не достаточно.
Используемый в настоящее время в практике морских исследований метод определения растворенного сероводорода в действительности позволяет определить суммарное содержание сернистых соединений (восстановленные формы серы), выражаемое в эквиваленте сероводорода [1, 2].
Метод количественного определения сероводорода основан на реакции окисления его иодом:
 . (1)
. (1)При этом 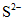 окисляется до свободной
окисляется до свободной  , а свободный иод переходит в ион
, а свободный иод переходит в ион  . Количественно эта реакция протекает лишь в кислой среде. В морской воде, обладающей слабощелочной реакцией, окисление иодом
. Количественно эта реакция протекает лишь в кислой среде. В морской воде, обладающей слабощелочной реакцией, окисление иодом  может идти дальше до образования сульфатного иона:
может идти дальше до образования сульфатного иона:
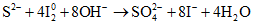 .
.Расход иода при этом окажется выше, чем необходимо для окисления растворенного сероводорода. Чтобы избежать этих ошибок, определение сероводорода ведут в кислой среде. При подкислении ионы 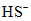 и
и  переходят в
переходят в 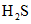 , и, таким образом, в подкисленной пробе весь содержащийся в морской воде сероводород будет находиться в виде слабодиссоциированной сероводородной кислоты
, и, таким образом, в подкисленной пробе весь содержащийся в морской воде сероводород будет находиться в виде слабодиссоциированной сероводородной кислоты  .
.
Для определения сероводорода к точно отмеренному раствору иода известной концентрации, подкисленному соляной кислотой, прибавляют определенное количество исследуемой морской воды. Раствор иода берется в избытке по отношению к ожидаемому содержанию сероводорода. Количество иода, израсходованного на окисление сероводорода, может быть легко определено по разности путем обратного титрования оставшегося иода раствором гипосульфита. Разница между количеством раствора гипосульфита, соответствующим всему количеству взятого для анализа иода, и количеством этого же раствора, затраченным на титрование избытка иода в пробе морской воды, будет эквивалентна содержанию сероводорода в исследуемой пробе.
Реакции между сероводородом и иодом и между иодом и гипосульфитом можно представить следующими уравнениями


В описываемом методе ошибки обусловлены следующими факторами: во-первых, растворы иода обладают значительной степенью летучести, их не следует оставлять открытыми на воздухе, а необходимо консервировать. Летучесть раствора иода снижают добавлением в него избытка иодида. При этом образуется трииодид-ион:
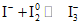 .
.Такие растворы уже значительно менее летучи. Если титрование происходит при температуре ниже 25 °C и содержание иодида в растворе иода составляет примерно 4%, то потеря иода в таких условиях ничтожно мала.
Во-вторых, иодиды в кислой среде окисляются кислородом воздуха:
 .
.Для того чтобы избежать этой ошибки, следует, по возможности, подкислять раствор иода, содержащий иодистый калий, непосредственно перед самым определением сероводорода и держать раствор в атмосфере углекислого газа.
Для выполнения анализа применяются:
бюретка для титрования по ГОСТ 20292;
пипетки 2-го класса точности на 1; 2; 5 и 10 мл по ГОСТ 20292;
колбы мерные 2-го класса точности на 200 и 250 мл по ГОСТ 1770;
колбы конические на 500 мл по ГОСТ 10394;
трубки стеклянные по ТУ 25-11-1045;
трубки резиновые по ГОСТ 5496;
склянки на 500 - 2000 мл по ТУ 6-19-6;
баллон со сжатой углекислотой с редукционным вентилем (возможно использование других источников углекислоты);
калий иодистый, х.ч., по ГОСТ 4232;
иод кристаллический, ч., по ГОСТ 4159;
натрий серноватистокислый, ч.д.а., по СТ СЭВ 223;
калий двухромовокислый, х.ч., по ГОСТ 4220;
калий иодноватокислый, х.ч., по ГОСТ 4202;
кислота соляная, х.ч., по ГОСТ 3118;
крахмал, ч., по ГОСТ 10163;
натрий углекислый кислый, ч.д.а., по ГОСТ 4201.
Заблаговременно до подъема на борт батометров с водой, в которой предполагается произвести определение сероводорода, приступают к подготовке колб для взятия проб воды.
По числу батометров, из которых будут взяты пробы на определение сероводорода, мерные колбы заполняют углекислотой по одному из способов, описанных ниже. После заполнения колб углекислотой калиброванной пипеткой отмеривают в каждую колбу раствор иода в иодистом калии концентрацией 0,01 моль/л. Раствор иода добавляют в избыточном количестве по отношению к растворенному в морской воде сероводороду. Например, для глубинных вод Среднего и Южного Каспия обычно бывает достаточно 1 мл раствора иода, для черноморской воды на глубине до 1000 м - 5 мл, ниже 1000 м - 10 мл. Колбы закрывают пробками до наполнения их морской водой. Колбы с раствором иода следует оберегать от нагревания и солнечного света.
Пробы для определения сероводорода отбирают после взятия проб для определения pH и содержания кислорода. Для этого резиновый шланг батометра промывают содержащейся в нем водой. При этом стеклянную трубку поднимают кверху для того, чтобы вытеснить пузырьки воздуха, которые иногда пристают к стенкам трубки или задерживаются в месте присоединения резинового шланга к крану батометра. После этого, не закрывая крана батометра, шланг зажимают пальцами, а стеклянную трубку, которой шланг оканчивается, опускают в колбу с раствором иода, непосредственно перед этим открытую. По мере заполнения колбы водой трубку постепенно поднимают и одновременно уменьшают скорость поступления воды, сжимая резиновый шланг пальцами; прекращают доступ воды, когда уровень точно достигнет метки на шейке колбы. В этот момент трубка наполнения должна быть выше черты мерной колбы. После заполнения колбу закрывают пробкой и переносят для дальнейшей обработки к титровальной установке. Жидкость в колбе должна сохранить желтый цвет иода; полное обесцвечивание жидкости по заполнении колбы указывает на недостаточное количество иода, взятого для определения. В этом случае проба должна быть взята повторно с большим количеством раствора иода в пробе.
1.4.1. Требования к посуде, применяемой в анализе
При определении сероводорода применяют тот же раствор гипосульфита (0,02 моль/л), что и при определении кислорода, поэтому следует пользоваться одной и той же бюреткой и титровальной установкой.
Для определения титра гипосульфита применяют ту же пипетку, что и при определении кислорода. Кроме того, необходимо иметь калиброванные пипетки для раствора иода. Мерные колбы используют с пришлифованными стеклянными или парафинированными корковыми пробками, свободно привязанными к каждой колбе. Колбы следует подобрать с приблизительно равными диаметрами шеек. Число колб зависит от числа проб морской воды, отбираемых одновременно для определения сероводорода. Все колбы должны быть пронумерованы.
1.4.2. Методы приготовления реактивов для проведения анализа
1. Раствор иода в иодистом калии концентрацией 0,01 моль/л готовят путем растворения 40 г химически чистого иодистого калия в 50 мл дистиллированной воды. В полученный раствор добавляют 2,54 г чистого кристаллического иода. По растворении кристаллов иода общий объем раствора доводят дистиллированной водой до литра. Иодистый калий должен быть испытан на чистоту по методу, описанному в гл. "Растворенный кислород". Раствор иода хранят в склянке оранжевого стекла или оклеенной черной фотографической бумагой с хорошо притертой пробкой.
Применение склянок с резиновыми пробками для хранения раствора иода не допускается.
2. Раствор гипосульфита концентрацией 0,02 моль/л готовят, как описано в гл. "Растворенный кислород".
3. Стандартные растворы двухромовокислого или иодноватокислого калия применяют те же, что и при определении кислорода.
4. Иодистый калий (проверенный на чистоту, см. гл. "Растворенный кислород").
5. Раствор соляной кислоты (1:1) готовят смешением одного объема химически чистой концентрированной соляной кислоты (плотность 1,19) с равным объемом дистиллированной воды (лить кислоту в воду, но не наоборот!).
6. Раствор крахмала применяют тот же, что и при определении кислорода.
7. Навески по 0,2 г бикарбоната натрия готовят заблаговременно взвешиванием 0,2 г соли на технических или ручных аптекарских весах и помещают в небольшие конвертики из кальки. Навески бикарбоната необходимы в том случае, если нет возможности пользоваться баллоном со сжатой углекислотой.
1.4.3. Определение поправки молярности раствора гипосульфита
Перед определением сероводорода находят поправочный коэффициент к концентрации 0,02 моль/л раствора гипосульфита и соотношению этого раствора с раствором иода (0,02 моль/л). Определение производят так же, как описано в гл. "Растворенный кислород". Результаты определения титра раствора гипосульфита со всеми поправками заносят в журнал.
1.4.4. Определение соотношения между раствором иода и раствором гипосульфита
Определение производят в тех же условиях, что и определение сероводорода в морской воде.
Мерную колбу (200 или 250 мл) заполняют углекислотой одним из двух следующих способов:
1. Если имеется баллон со сжатой газообразной углекислотой, то чистую, сполоснутую дистиллированной водой колбу наполняют в течение нескольких секунд углекислотой из баллона. Наполнение производят через стеклянную опущенную до дна колбы трубку, соединенную с редукционным вентилем баллона резиновым шлангом. Даже при слабой струе газа для наполнения колбы углекислотой достаточно нескольких секунд.
2. Если для заполнения колб углекислотой применяют навески бикарбоната натрия, то поступают следующим образом: в чистую, сполоснутую дистиллированной водой колбу добавляют 2 мл соляной кислоты (1:1), затем всыпают 0,2 г бикарбоната натрия. По растворении навески колба наполняется углекислотой, выделившейся при разложении бикарбоната натрия соляной кислотой.
После заполнения колбы углекислотой в нее добавляют 10 мл раствора иода (0,02 моль/л) и, если наполнение колбы углекислотой производилось из баллона, прибавляют 1 мл разведенной (1:1) соляной кислоты. Колбу тотчас закрывают пробкой. Промывалку, наполненную поверхностной бессероводородной морской водой, соединяют резиновым шлангом со стеклянной трубкой. Наклоняя промывалку с водой, заполняют шланг и трубку водой так, чтобы в них не оставалось пузырьков воздуха. Затем, придерживая резиновый шланг пальцами, опускают стеклянную трубку в колбу и, покачивая колбу, постепенно заполняют ее водой. По мере заполнения колбы стеклянную трубку поднимают, одновременно уменьшают скорость поступления воды, сжимая шланг пальцами, и осторожно доводят объем воды до метки колбы. Колбу после заполнения закрывают стеклянной пробкой и содержимое перемешивают перевертыванием колбы (придерживая пробку). Содержимое мерной колбы переливают в коническую колбу, и иод титруют гипосульфитом, как это описано в гл. "Растворенный кислород". При окислении сероводорода иодом выделяется тонкодисперсная сера, титруемая жидкость опалесцирует, принимает слегка желтоватую окраску, поэтому во избежание визуальных ошибок крахмал следует добавлять при отчетливом желтом цвете титруемой жидкости. В противном случае проба может быть легко перетитрована.
В конце титрования часть титруемого раствора переливают в мерную колбу, в которую наливалась бессероводородная морская вода. Мерную колбу ополаскивают раствором и снова сливают в титровальную колбу. Вновь посиневшую жидкость осторожно дотитровывают, после чего производят отсчет по шкале (m) бюретки и записывают в журнал (табл. 12).
Форма журнала для записи результатов определения H2S
Установка титра раствора гипосульфита: дата 5.01.89 г., время 11.00, стандарт KIO3. Молярная концентрация стандарта 0,02 моль/л. Объем пипетки (марка) 10,00 мл, поправка 0,03, истинный объем 10,03. Расход гипосульфита при установке титра на 10,03 мл стандарта - ...
Поправка раствора гипосульфита K = 0,985 | Отсчеты | Среднее значение | |
I. 10, 14 | отсчет ... 10,16 | ||
II. 10, 18 | поправка ... 0,02 | ||
исправленный отсчет ... 10,18 | |||
Определение соотношения растворов гипосульфита и иода | |||
Отсчет по бюретке m | ... 9,95 | ||
Поправка | ... 0,03 | ||
Исправленный отсчет n | ... 9,98 | ||
N п/п | N станции | Дата и время | Горизонт, м | N колбы | Объем колбы, V | V - d | Коэффициент A | Отсчет бюретки n1 | Поправка на бюретку | Исправленный отсчет n | m - n | MK | MK x (m - n), [H2S], мл/л |
1 | 26 | 05.01.89 г. 9.00 | 1000 | 250 | 237,97 | 0,929 | 6,83 | +0,03 | 6,86 | 3,12 | 0,915 | 2,85 |
Если титруемая жидкость не посинела после сливания раствора, которым ополаскивалась мерная колба, то это указывает на то, что проба перетитрована.
Всю операцию по определению отношения раствора иода и гипосульфита производят дважды. Расхождения результатов параллельных определений не должны превышать 0,05 - 0,1 мл.
Титрование проб следует производить непосредственно после взятия пробы. Длительное стояние проб ведет к потерям иода и дает значительные погрешности.
Для титрования пробу переливают из мерной колбы в коническую и титруют раствором гипосульфита, как это описано выше, при определении соотношения растворов иода и гипосульфита. Результат отсчета по шкале бюретки записывают в журнал (см. табл. 12).
Содержание сероводорода в морской воде принято выражать в миллилитрах газообразного H2S (приведенного к t = 0 °C и P = 760 мм рт. ст.) в литре воды.
Из уравнений реакции между иодом и сероводородом (2) и между иодом и гипосульфитом (3) следует, что 1 г/моль сероводорода соответствует 2 г/моль гипосульфита. Относительная молекулярная масса сероводорода равна 34,08, а масса 1 мл при 0 °C и 760 мм рт. ст. составляет 1,5393 мг. Исходя из этих соотношений, содержание сероводорода (мл/л) вычисляют по формуле

где 34 - приближенное значение относительной молекулярной массы сероводорода; m - объем раствора гипосульфита, затраченного на титрование иода при определении соотношения растворов иода и гипосульфита, мл; n - объем раствора гипосульфита, затраченного на титрование пробы, мл; M - молярная концентрация раствора гипосульфита; K - поправочный коэффициент раствора гипосульфита; V - объем колбы; d - объем добавленных в колбу реактивов (раствора иода, соляной кислоты).
Если при определении всегда применяется раствор гипосульфита концентрацией 0,02 моль/л, то формула (4) может быть упрощена. Поскольку
 ,
,тогда:

Если для определения H2S применяются одни и те же колбы и одно и то же количество вводимых в колбу реактивов, целесообразно для каждой колбы вычислить коэффициент
 ,
,а значения коэффициента A для каждой серии колб свести в особую таблицу; тогда содержание сероводорода (мл/л) составит:
 . (6)
. (6)Пример.
1. При определении титра гипосульфита найдено, что на 10,03 мл (истинный объем пипетки) стандартного раствора иодата калия KIO3 концентрацией 0,0033 моль/л израсходовано 10,18 мл раствора гипосульфита. (Среднее из двух определений с учетом поправки бюретки: b1 = 10,14; b2 = 10,18; bср = 10,16. Соответствующая поправка бюретки 0,02, тогда b = 10,18.) Тогда молярная концентрация раствора гипосульфита
 ,
,где K = 0,985; 6 - стехиометрический коэффициент реакции.
2. При нахождении соотношения между растворами иода и гипосульфита взято 10,03 мл раствора иода и 2 мл раствора соляной кислоты. На титрование этого объема иода израсходовано 9,98 мл (с учетом поправки на калибрацию бюретки) раствора гипосульфита концентрацией 0,02 моль/л. Для определения применялась колба емкостью 250 мл.
3. При определении сероводорода в пробе взяты те же объемы иода и соляной кислоты. На титрование иода, оставшегося после окисления сероводорода, израсходовано 6,86 мл раствора гипосульфита концентрацией 0,02 моль/л (с учетом поправки на калибрацию бюретки).
4. Подставляя эти значения в формулу (5), находим:
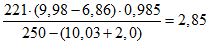 (мл/л). (7)
(мл/л). (7)5. Если предварительно вычислено значение

то содержание сероводорода составит
0,929 (9,98 - 6,86) 0,985 = 2,85 (мл/л).
Очевидно, что вычисление по формуле (8) проще и занимает меньше времени, что имеет существенное значение при обработке результатов наблюдений в экспедиционных условиях - на борту судна.
Содержание сероводорода вычисляют с точностью до двух десятичных знаков. Результаты определений и вычислений заносят в журнал (см. табл. 12).
Определения сероводорода может проводить техник или старший техник-химик, знакомый с основами объемного химического анализа.
Для анализа 100 проб требуется 22 чел.- ч, в том числе:
на взятие проб из батометра - 2,0 чел.-ч;
на приготовление растворов реактивов - 3,5 чел.-ч;
на подготовку посуды - 3,2 чел.-ч;
на выполнение измерений - 7,5 чел.-ч;
на выполнение расчетов - 5,8 чел.-ч.
--------------------------------
<*> Настоящая методика метрологически не аттестована.
Метод основан на реакции сульфид-ионов подкисленной пробы морской воды с N,N-диметил-n-фенилендиамином (диамином) в присутствии ионов железа (III) как катализатора. В процессе реакций окисления и замещения происходит количественное включение сульфидной серы в гетероцикл красителя - метиленового синего. Полученные окрашенные пробы колориметрируют относительно холостой пробы при длине волны 670 нм в 1-, 5- или 10-сантиметровых кюветах в зависимости от концентраций сероводорода [1, 3, 4].
Для выполнения анализа применяются:
спектрофотометр, позволяющий производить измерения при длине волны 670 нм, по ТУ 3-3-1314;
или фотометр с фильтром, близким к 670 нм, по ТУ 3-3-748;
батометр пластмассовый или стеклянный ГР-18 по ТУ 25-04-2507;
пипетки автоматические по ГОСТ 20292;
цилиндры Несслера по ТУ 25-11-1023;
колба круглодонная одногорлая на 1 л по ГОСТ 10394;
колбы мерные на 0,5 и 1,0 л по ГОСТ 1770;
бюксы диаметром 50 - 80 мм по ГОСТ 7148;
плитка электрическая бытовая по ТУ 92-208;
пипетки с делениями по ГОСТ 20292;
N,N-диметил-n-фенилендиамин дигидрохлорид, ч.д.а., по ТУ 6-09-1903;
железо хлорное, ч.д.а., по ГОСТ 4147;
кислота серная, х.ч., по ГОСТ 4204;
натрий сернистый, ч.д.а., по ГОСТ 2053;
натрий серноватистокислый, ч.д.а., по СТ СЭВ 223;
калий иодноватокислый, х.ч., по ГОСТ 4202;
калий иодистый, х.ч., по ГОСТ 4232;
крахмал, ч., по ГОСТ 10163;
спирт изобутиловый, ч., по ГОСТ 6016;
спирт этиловый, х.ч., по ТУ 6-09-1710;
кислота соляная, х.ч., по ГОСТ 3118;
азот газообразный, ос.ч., по ГОСТ 9293.
Пробы морской воды на сероводород отбирают пластмассовым батометром сразу после отбора проб на кислород. Хранение проб не допускается.
2.4.1. Методы приготовления реактивов для проведения анализа
1. Раствор N,N-диметил-n-фенилендиамина готовят растворением 1 г реактива в 500 мл соляной кислоты концентрацией 6 моль/л.
2. Раствор соляной кислоты (6 моль/л) готовят разбавлением концентрированной соляной кислоты (37%, плотность 1,19 г/см3) равным объемом дистиллированной воды.
3. Раствор хлорного железа готовят растворением 8 г соли в соляной кислоте концентрацией 6 моль/л с последующим доведением объема до 500 мл.
4. Бескислородную воду получают кипячением в течение 30 мин дистиллированной воды. В процессе кипячения через воду продувают газообразный азот. После охлаждения воду постоянно продувают азотом в течение всего времени, пока вода необходима для приготовления градуировочных растворов.
5. Раствор серной кислоты (1:4) готовят добавлением 1 объема кислоты к 4 объемам воды.
6. Основной раствор сульфида натрия готовят растворением навесок (0,750 г) Na2S в бескислородной воде и доведением объема до 1000 мл в мерной колбе. Полученный раствор содержит около 3,12 мкмоль/мл сульфид-иона. Перед взвешиванием кристаллы сульфида натрия быстро промывают дистиллированной водой, высушивают на фильтровальной бумаге и хранят в герметически закрытых бюксах.
7. Рабочий раствор сульфида натрия с концентрацией около 0,156 мкмоль/мл  готовят разбавлением 25 мл основного раствора сульфида натрия бескислородной водой до 500 мл. Раствор устойчив 15 - 30 мин.
готовят разбавлением 25 мл основного раствора сульфида натрия бескислородной водой до 500 мл. Раствор устойчив 15 - 30 мин.
8. Раствор тиосульфата натрия концентрацией 0,02 моль/л готовят растворением 5 г тиосульфата натрия в 1000 мл дистиллированной воды. Для стабилизации раствора добавляют 5 мл изобутилового спирта.
9. Раствор иодата калия готовят растворением 1,1891 г соли, высушенной в течение часа при 180 °C, в 1000 мл дистиллированной воды.
10. Иодистый калий очищают от свободного иода промыванием соли спиртом до появления бесцветной порции спирта.
11. Раствор крахмала готовят растворением 1 г крахмала в 100 мл дистиллированной воды.
2.4.2. Стандартизация раствора тиосульфата натрия
Вследствие неустойчивости раствора тиосульфата натрия необходимо периодически определять поправочный коэффициент для его концентрации.
В коническую колбу после растворения 1 г иодида калия в 40 - 50 мл дистиллированной воды вносят 2 мл серной кислоты. Затем пипеткой прибавляют 15 мл раствора иодата калия концентрацией 0,02 моль/л, колбу закрывают, перемешивают и после выдерживания раствора в течение минуты приступают к титрованию. До появления светло-желтой окраски раствора титрование проводят без индикатора, затем прибавляют 1 мл раствора крахмала и 50 мл дистиллированной воды и продолжают титрование до полного обесцвечивания раствора. Опыт повторяют 2 - 3 раза.
Поправочный коэффициент для концентрации раствора тиосульфата натрия вычисляют по формуле
К = а / b,
где: a - истинный объем калиброванной пипетки; b - истинный отсчет на калиброванной бюретке.
2.4.3. Стандартизация рабочего раствора сульфида натрия
Операция проводится в течение нескольких минут после приготовления рабочего раствора и одновременно с приготовлением градуировочных растворов для фотометрирования.
В шесть колб с притертыми пробками приливают по 10 мл дистиллированной воды и добавляют по 1 - 2 г иодида калия. В каждую колбу добавляют 10,00 мл раствора иодата и 1,0 мл серной кислоты. Затем в три колбы вносят по 50 мл рабочего раствора сульфида, а в остальные - по 50 мл дистиллированной воды. Выдерживают все колбы в прохладном месте, а затем титруют содержимое колб раствором тиосульфата, используя в качестве индикатора крахмал.
Расчет проводят по формуле
 ,
,где A - среднее значение, полученное по результатам трех титрований проб без добавки сульфид-иона, мл; B - среднее значение, полученное по результатам трех титрований проб с добавкой сульфид-иона, мл; M - концентрация раствора тиосульфата, моль/л.
Расхождение между результатами трех титрований не должно превышать 0,05 мл.
2.4.4. Фотометрирование градуировочных растворов
Из рабочего раствора готовят серию градуировочных растворов. Для этого в мерные колбы на 100 мл добавляют следующие объемы рабочего раствора: 0; 4; 8; 12; 16; 20 мл, получая таким образом растворы с концентрациями сульфид-иона 0,0; 6,3; 12,5; 18,7; 25,0 и 31,2 мкмоль/л, в том случае, если рабочий раствор сульфид-иона имеет концентрацию точно 0,156 мкмоль/мл. Поэтому полученные концентрации градуировочных растворов должны быть скорректированы с учетом истинной концентрации рабочего раствора сульфид-иона, определенной титрованием.
Затем колбы заполняют бескислородной водой до отметки 100 мл. С помощью автоматической пипетки в каждую колбу вносят по 1 мл раствора диамина и хлорного железа и содержимое тщательно перемешивают. Через 60 мин измеряют оптические плотности растворов относительно холостой пробы (бескислородная вода с реактивами) при длине волны 670 нм в 1-, 5- или 10-сантиметровых кюветах. По полученным результатам строят градуировочные графики для каждого вида кювет. Градуировочный график представляет собой прямую линию, проходящую через начало координат.
При анализе более высоких концентраций начиная с 40 - 50 мкмоль/л градуировочный график отклоняется от прямой. Точка отклонения графика от прямой зависит от качества раствора N,N-диметил-n-фенилендиамина.
Пробы на сероводород отбираются аналогично пробам на кислород. Сразу после отбора в пробы добавляют по 1 мл растворов диамина и хлорного железа - предпочтительно автоматическими пипетками. Закрывают склянки пробками и тщательно перемешивают растворы. Голубая окраска начинает проявляться через несколько минут, и к фотометрированию проб можно приступить через 30 мин. Однако, если пробы содержат высокие концентрации сероводорода, то для полного окрашивания необходимо 60 мин. Окраска устойчива по меньшей мере в течение 24 ч, ее интенсивность измеряют относительно дистиллированной воды (или, если необходимо при низких концентрациях сероводорода, относительно фона реактивов) при длине волны 670 нм, используя 1-, 5- или 10-сантиметровые кюветы.
Если пробы содержат более 100 мкмоль/л сероводорода, их следует разбавить перед анализом. При этом нужный объем пробы помещают в мерную колбу, содержащую бескислородную воду. Носик пипетки следует погрузить ниже поверхности воды. Затем объем доводят до метки бескислородной водой. Вводят необходимые реактивы и пробу тщательно перемешивают. При расчете результатов анализа необходимо учесть фактор разбавления.
Иногда может возникнуть необходимость компенсировать оптическую плотность применяемых реактивов. Фон реактивов получают, добавляя их к фильтрованной поверхностной морской воде и измеряя оптическую плотность относительно той же морской воды без реактивов. Полученные значения не должны превышать 0,5 в 10-сантиметровой кювете (желательно, чтобы они были ниже 0,25). Обычно фон реактивов незначителен, даже если используют окрашенный раствор диамина.
Примечание. Трудно приготовить стандартный раствор сульфида с достаточной степенью точности. Таким образом, в анализе появляется систематическая ошибка. В описываемом методе эта ошибка ниже 2%.
Для рутинного анализа используют калибровочный фактор. Для этого с помощью градуировочного графика по измеренному значению оптической плотности (например, 0,500) находят соответствующее значение концентрации (например, 20,2 мкмоль/л) и рассчитывают фактор F для данной кюветы, которую затем используют для измерений:
 , т.е. F = 40,4.
, т.е. F = 40,4.Затем получают концентрацию в пробе (мкмоль/л), умножая оптическую плотность пробы на значение F.
Примечание. Трудно достать реактив диамина, который был бы в большей или меньшей степени окрашен. Однако это не влияет на результаты. Исследования, проведенные с коричневым реактивом диамина годичной давности, дали результаты, лишь незначительно отличающиеся от полученных со свежеприготовленным раствором диамина.
Анализ может выполнять химик-аналитик, знакомый с основами объемного химического анализа и с правилами эксплуатации приборов, применяемых в данной методике.
Для анализа 100 проб требуется 22,2 чел.-ч, в том числе:
на взятие проб из батометра - 2,5 чел.-ч;
на подготовку посуды - 3,2 чел.-ч;
на приготовление растворов реактивов - 3,5 чел.-ч;
на подготовку спектрофотометра к работе - 1 чел.-ч;
на выполнение измерений - 10 чел.-ч;
на выполнение расчетов - 2 чел.-ч.
3. Chemical methods for use in marine environmental monitoring/IOC, Manuals and guides, N 12. UNESCO, 1983, p. 11 - 16.
4. Methods of seawater analysis/Grasshoff K. et al. (Eds.). Verlag Chemie, Weinheim, 1983, p. 73 - 80.
ФОТОМЕТРИЧЕСКИМИ МЕТОДАМИ
Биогенные вещества (фосфаты, нитраты, нитриты, аммонийный азот, кремний) являются важнейшими ингредиентами природных вод. В устьевых областях рек в большинстве случаев наблюдаются повышенные концентрации этих веществ, что вызывает интенсивное развитие фитопланктона.
Для определения биогенных веществ в природных водах применяют фотометрические методы [1, 2]. Практика показывает, что при анализе распресненных вод морских устьевых областей рек с высоким содержанием взвешенных частиц минерального и органического происхождения удовлетворительной воспроизводимости результатов определения биогенных веществ можно достигнуть только при условии предварительного фильтрования проб для отделения взвеси. В открытых частях эпиконтинентальных морей, где содержание взвешенных частиц незначительно, фильтрование проб можно не производить.
1.1. При определении фосфатов и общего фосфора в распресненных водах любой солености, а также нитритов и нитратов в водах с соленостью до 7+ применяются средства измерения, оборудование и материалы, указанные в [1] и [2].
1.2. При определении кремния и аммонийного азота в распресненных водах любой солености, а также нитритов и нитратов в водах с соленостью выше 7+ применяются средства измерения, оборудование и материалы, указанные в [1].
Пробы воды отбирают металлическими батометрами БМ-48 и сразу же фильтруют через ядерный или мембранный фильтр с размером пор 0,45 мкм с применением фильтровальной установки любого типа, например так, как указано в [1] в п. "Определение хлорофилла и феофитина". Прозрачные пробы не фильтруют. Пробы воды не консервируют, анализ необходимо производить по возможности сразу после отбора. Допускаемые сроки хранения проб указаны в [1].
3.1. Подготовительные операции и ход определения фосфатов и общего фосфора в распресненных водах любой солености, а также нитритов и нитратов в водах с соленостью до 7+ производятся так, как указано в [1] и [2].
3.2. Подготовительные операции и определение кремния и аммонийного азота в распресненных водах любой солености, а также нитритов и нитратов в водах с соленостью выше 7+ производятся так, как указано в [1].
При определении низких концентраций фосфатов и нитритов рекомендуется применять кюветы длиной 100 мм. Лучшие результаты могут быть достигнуты при применении спектрофотометра (типа СФ-4А, СФ-16, СФ-26, СФ-46, "Спекол" и др.). В судовых условиях удобнее пользоваться фотоэлектроколориметрами с цифровой шкалой (ФЭК, КФК) или спектрофометром "Спекол". Работать с кюветами меньшей длины можно, начиная с концентраций фосфатов выше 10 мкг/л, нитритов (и соответственно нитратов) - выше 4,9 мкг/л.
Несоблюдение указанных требований приводит к резкому ухудшению точности определений.
Кроме того, как указано выше, пробы распресненных вод, отобранных в устьевых областях рек, должны быть предварительно отфильтрованы для отделения взвеси.
Раздел исключен с 1 июля 2011 года. - РД 52.10.738-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.739-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.744-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.740-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2011 года. - РД 52.10.745-2010, утв. Росгидрометом.
Раздел исключен с 1 июля 2014 года. - РД 52.10.772-2013, утв. Росгидрометом 29.09.2013.
Методика позволяет определять общий и органический азот в морских и распресненных водах, предназначена для проведения мониторинга этих вод и характеризуется пределом обнаружения азота около 30 мкг/л. Диапазон определяемых концентраций общего и органического азота - 30 - 5000 мкг/л. Анализу не мешают любые ионы или соединения, присутствующие в чистых или умеренно загрязненных морских водах. Также не мешает определению сероводород при концентрациях до 2 мг/л. При анализе же вод Черного моря, в которых содержание сероводорода может доходить до 20 мг/л, пробы следует разбавить безазотной водой до указанной выше концентрации.
Азот в морской воде входит в состав как неорганических соединений (нитриты, нитраты, соли аммония), так и органических (гуминовые и фульвовые вещества, белки, аминокислоты, амины, амиды и др.). Эти соединения относятся к числу важнейших биогенных веществ, в значительной степени определяющих биологическую продуктивность морей и океанов. Изменения в составе форм азота указывают на направление основных биохимических и гидробиологических процессов в морской среде.
Многообразие форм азота в морской воде исключает прямые методы определения его общего содержания. Нашедшие практическое применение методы определения общего азота основаны на переводе органических соединений в неорганические: аммиак (метод Кьельдаля), нитраты и нитриты (фотохимическое и персульфатное окисление).
Классический метод Кьельдаля в его различных вариантах [2, 3] дает надежные результаты только при отсутствии высоких концентраций нитритов и нитратов. Его существенными недостатками являются также сложность и трудоемкость аналитических процедур. Более простыми и в то же время не менее чувствительными являются способы фотохимического [2, 4, 7] и персульфатного [5, 6] окисления азоторганических соединений до нитратов и нитритов. Они позволяют определять общий азот по одной пробе, при этом первый из них - только в растворе, а второй - в растворе и во взвеси, что делает последний более предпочтительным. Ниже описан метод определения общего (и органического) азота в морской воде с применением персульфатного окисления в щелочной среде [1, 5, 6].
Метод основан на окислении азоторганических соединений и аммонийного азота до нитратов и нитритов при кипячении с персульфатом калия в щелочной среде. Сумму нитратов и нитритов восстанавливают омедненным кадмием до нитритов и количественно определяют с помощью реактива Грисса-Илосвая. Метод достаточно прост и может применяться на современных научно-исследовательских судах [1].
Для выполнения анализа применяются:
спектрофотометр СФ-26 (СФ-16, СФ-4А) или фотоэлектроколориметр ФЭК-56 (ФЭК-56М, КФК-3, ФЭК-60) с кюветами длиной 20 и 50 мм;
батометр, например ГР-18, по ТУ 25-04-2507;
кастрюля-скороварка объемом 6 л;
бутылки для автоклавирования на 50 мл, закрытые пробками, со скобяным замком и прокладками из силиконовой резины или кислородная склянка с хорошо притертой пробкой;
плитка электрическая бытовая ПЭК-800/3 по ТУ 92-208;
стеклянный аппарат для перегонки воды любого типа;
пипетки с делениями на 1; 2; 5 и 10 мл по ГОСТ 20292;
пипетка автоматическая калиброванная на 10 мл;
пипетка калиброванная на 5 мл по ГОСТ 20292;
колбы мерные по ГОСТ 1770 или цилиндры Несслера на 100 мл с притертыми пробками;
склянки для хранения растворов из темного стекла и пластмассовыми пробками на 1 л;
фильтры мембранные N 2 с размером пор 0,45 мкм и диаметром 35 мм;
фильтр стеклянный N 2;
колба Бунзена на 0,5 л;
насос водоструйный стеклянный по ГОСТ 10696 или пластмассовый КМ-1230 по ТУ 64-1-861, или механический Комовского типа НВК;
стакан химический на 0,4 - 0,6 л по ГОСТ 10394;
стекла часовые;
бюксы низкие диаметром 40 - 50 мм по ГОСТ 7148;
серная кислота концентрированная, х.ч., по ГОСТ 4204;
натрия гидроксид, х.ч., по ГОСТ 4328;
калий надсернокислый (калий персульфат), х.ч., по ГОСТ 4146;
борная кислота, х.ч., по ГОСТ 9656;
трилон Б (этилендиамин-N,N,N',N'-тетрауксусной кислоты динатриева соль двухводная), ч., по ГОСТ 10652.
Пробы морской воды на общий азот анализируют сразу же после их отбора. Хранение проб не допускается. Если пробы морской воды содержат большое количество взвешенных частиц, что характерно для предустьевых районов морей, то до анализа их следует профильтровать через мембранный фильтр N 2 по [3].
4.1.1. Раствор гидроксида натрия концентрацией 0,12 моль/л готовят растворением 4,8 г щелочи в 1 л дистиллированной воды. Хранят в склянке с пластмассовой пробкой.
4.1.2. Раствор гидроксида натрия концентрацией 0,075 моль/л готовят растворением 3,02 г щелочи в 1 л безазотной воды.
4.1.3. Боратный буфер готовят растворением 6,0 г борной кислоты в 1 л раствора гидроксида натрия концентрацией 0,075 моль/л. Он устойчив на холоде не менее двух месяцев.
4.1.4. Окислительный раствор готовят растворением 10,0 г персульфата калия в 1 л боратного буфера. При хранении в темной склянке с пластмассовой пробкой на холоде раствор устойчив по крайней мере неделю.
В химический стакан на каждые 10 фильтров приливают 50 мл безазотной воды. Закрывают его часовым стеклом, ставят на электроплитку, покрытую асбестовой тканью, и нагревают при слабом кипении раствора в течение 20 мин три раза, каждый раз меняя воду. Фильтры хранят в бюксе.
Безазотная вода. 1 л дистиллированной воды помещают в колбу перегонного аппарата, добавляют 0,5 г персульфата калия и 50 мл раствора гидроксида натрия концентрацией 0,12 моль/л, нагревают раствор до кипения на газовой горелке и кипятят несколько минут без отгонки. Затем собирают перегонный аппарат и отгоняют до тех пор, пока в колбе не останется 150 мл раствора. На безазотной воде готовят все реактивы и стандартные растворы. Воду желательно получать в день употребления.
Допускается ее хранение в течение 3 - 4 недель в склянках или колбах с хорошо пришлифованными и запарафинированными пробками в помещениях, воздух которых свободен от паров аммиака, азотной кислоты и органических соединений азота.
Большое внимание следует уделять дистиллированной воде, которая нередко так сильно бывает загрязнена органическим азотом, аммиаком, нитритами и нитратами, что, с одной стороны, полученная из нее безазотная вода также может содержать повышенные концентрации соединений азота, а с другой стороны, сильно загрязняет нитритами и нитратами пробы природной воды и холостую пробу при разбавлении. Для уменьшения содержания соединений азота дистиллированную воду следует получать в день использования.
Безазотная и дистиллированная воды пригодны для анализа, если в кювете длиной 50 мм оптическая плотность холостой пробы, измеренная относительно дистиллированной воды, не превышает 0,060. При больших значениях оптической плотности вместо дистиллированной воды следует использовать безазотную воду. Если же в безазотной воде содержание соединений азота повышено, то ее необходимо еще раз перегнать с персульфатом калия в щелочной среде.
Деионизированную воду применять не следует, так как она обычно содержит довольно много органического азота.
Альфа-нафтиламин. Этим реактивом можно пользоваться в том случае, если в твердом состоянии он окрашен в слабо-розовый цвет. При более интенсивной окраске его очищают. Для этого 0,2 г альфа-нафтиламина растворяют в 40 мл безазотной воды и нагревают до кипения. Горячий раствор фильтруют через фильтр из обычной фильтровальной бумаги, вставленной в стеклянную химическую воронку, предварительно нагретую в сушильном шкафу. В фильтрованный раствор добавляют 15 мл ледяной уксусной кислоты и 245 мл безазотной воды.
Калий надсернокислый. Его очистка описана в гл. "Общая растворенная ртуть".
Воздух лабораторного помещения, в котором проводят анализ на общий и органический азот, а также получают дистиллированную и безазотную воды, должен быть чистым и свободным от паров азотной кислоты, аммиака и органических производных азота даже в следовых количествах. Этого можно достигнуть только в том случае, если в данном помещении не работали с указанными выше соединениями азота не менее шести месяцев. Также недопустимо присутствие в воздухе табачного дыма. В противном случае лабораторное помещение "отравлено" соединениями азота, и результаты анализов плохо воспроизводятся.
В зависимости от ожидаемой концентрации общего азота в пробе отбирают 10 мл (концентрация не больше 1000 мкг/л) или 2 мл (больше 1000 мкг/л) морской воды в бутылки для автоклавирования, прибавляют 10 мл окислительного раствора, бутылки сразу же закрывают и их содержимое перемешивают. Бутылки помещают в кастрюлю-скороварку, в которую предварительно наливают 600 мл дистиллированной воды. Кастрюлю закрывают и ставят на электроплитку. Бутылки с растворами кипятят 30 мин при 120 °C с работающим грузовым клапаном (от начала нагревания до конца кипячения обычно требуется около 70 мин), после чего кастрюлю снимают с плитки. После охлаждения кастрюлю открывают и бутылки вынимают. Содержимое бутылок количественно переносят в цилиндры Несслера на 100 мл и доводят дистиллированной водой до метки. Полученные растворы анализируют далее по методике определения нитратов в морской воде с той лишь разницей, что после пропускания через колонку с омедненным кадмием отбирают по 25 мл пробы и по 25 мл (при измерении на спектрофотометре) или 50 мл (при измерении на фотоэлектроколориметре) холостой пробы. Необходимо отметить, что для вод разной солености следует прибавлять различные объемы щелочного раствора трилона Б для создания оптимального значения pH, которое надо строго контролировать. Параллельно с пробами каждый раз обрабатывают в кастрюле-скороварке окислительный раствор (холостую пробу) с последующим его разбавлением дистиллированной водой до 100 мл.
Для выполнения холостого определения 10 мл окислительного раствора проводят через все стадии анализа, предусмотренные для пробы.
Основной стандартный раствор трилона Б готовят растворением 0,1330 г соли в безазотной воде в мерной колбе на 100 мл. 1 мл этого раствора содержит 100 мкг органического азота. При хранении в холодном месте в темной посуде раствор устойчив несколько месяцев.
Рабочий стандартный раствор трилона Б готовят разбавлением 10,0 мл основного стандартного раствора безазотной водой в мерной колбе на 100 мл до метки. 1 мл этого раствора содержит 10 кг органического азота. При хранении в холодном месте раствор устойчив месяц.
Поскольку концентрация общего азота в морской воде может изменяться от сотен до нескольких тысяч микрограммов на литр, градуировочные графики необходимо строить в диапазонах концентраций 0 - 1000 или 0 - 5000 мкг/л.
Для построения градуировочного графика в диапазоне 0 - 1000 мкг/л отбирают 2,5; 5,0; 7,5; 10,0 мл рабочего стандартного раствора трилона Б и разбавляют безазотной водой в мерных колбах или цилиндрах Несслера на 100 мл до метки. Концентрация азота в полученных стандартных растворах составляет соответственно 250; 500; 750 и 1000 мкг/л.
Отбирают пипеткой и помещают в бутылки для автоклавирования по 10 мл каждого стандартного раствора, добавляют к ним по 10 мл окислительного раствора и сразу же закрывают бутылки пробками. После перемешивания растворов бутылки (до 8 - 10 шт.) помещают в кастрюлю-скороварку и далее проводят аналитические процедуры, аналогичные описанным в п. 5.1.
Добавляют 2,5 и 1,25 мл реактива Грисса-Илосвая к 50 и 25 мл раствора соответственно и затем измеряют оптическую плотность градуировочных растворов на спектрофотометре при длине волны 543 нм или на фотоэлектроколориметре при длине волны, наиболее близкой к указанной, в кюветах длиной 50 мм относительно холостой пробы.
Каждый стандартный раствор готовят параллельно не менее трех раз. Градуировочный график строят по средним измеренным значениям оптической плотности растворов в координатах "оптическая плотность - концентрация общего азота, мкг/л".
Для построения градуировочного графика в диапазоне концентраций азота 0 - 5000 мкг/л отбирают по 1,0; 2,0; 3,0; 4,0; 5,0 мл основного стандартного раствора трилона Б и разбавляют их безазотной водой в мерных колбах или цилиндрах Несслера на 100 мл до метки. Полученные стандартные растворы имеют концентрации азота 1000; 2000; 3000; 4000; 5000 мкг/л соответственно. Отбирают пипеткой в бутылки для автоклавирования по 2 мл каждого раствора, добавляют к ним по 10 мл окислительного раствора и сразу же закрывают пробками. Дальнейший ход анализа и построение градуировочного графика проводят аналогично описанному для диапазона концентраций 0 - 1000 мкг/л. Следует отметить нестабильность окраски растворов при концентрациях общего азота более 3000 мкг/л. Поэтому необходимо строго соблюдать время выдержки проб от момента внесения реактива Грисса-Илосвая до измерения оптической плотности и выполнять анализ сериями не более 10 проб.
Градуировочные графики следует проверять не реже одного раза в месяц и обязательно каждый раз при приготовлении новых растворов реактивов.
Оптическую плотность проб измеряют в кювете длиной 50 мм относительно холостой пробы на спектрофотометре при длине волны 543 нм или фотоэлектроколориметре со светофильтром N 6 (ФЭК-56М, ФЭК-56) или N 4 (ФЭК-60) и записывают ее в журнал. Когда оптическая плотность определяемой пробы в диапазоне концентраций 0 - 5000 мкг/л превышает 0,8 в кювете длиной 50 мм, необходимо провести ее повторное измерение в кювете длиной 20 мм.
По измеренным значениям оптической плотности с помощью градуировочных графиков находят концентрацию общего азота (мкг/л). Если измерение оптической плотности проведено в кюветах длиной 20 мм, то в этом случае найденную по градуировочному графику концентрацию общего азота следует умножить на 2,5 для получения его истинной концентрации.
Если ранее в пробе были определены концентрации нитритов, нитратов и аммонийного азота, то, вычитая их сумму из найденной концентрации общего азота, находят содержание в морской воде органического азота (в мкг/л).
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09.88 по 25.12.88 (табл. 19), настоящая методика определения общего и органического азота допущена к применению в организациях Росгидромета.
Таблица 19
Результаты метрологической аттестации МВИ
Диапазон концентрации общего азота в морской воде, мкг/л | Показатель воспроизводимости ( | Показатель правильности ( | Показатель погрешности МВИ, суммарная погрешность ( |
250 - 500 | 4,5 | 10,5 | 12,0 |
500 - 750 | 3,5 | 9,5 | 9,5 |
750 - 2600 | 1,2 | 4,6 | 4,6 |
Необходимая точность определения общего азота может быть достигнута при правильном контроле чистоты безазотной воды, реактивов, посуды и воздуха лабораторного помещения.
Определение может выполнять химик-аналитик, имеющий среднее специальное образование.
Для анализа общего азота в 10 пробах требуется 19,8 чел.-ч, в том числе:
на взятие проб из батометра - 0,3 чел.-ч;
на подготовку посуды и приготовление растворов реактивов - 2,0 чел.-ч;
на подготовку очищенной воды - 6,0 чел.-ч;
на подготовку редукторов - 3,0 чел.-ч;
на фильтрование проб - 2,0 чел.-ч;
на выполнение измерений - 3,5 чел.-ч;
на измерение светопоглощения - 0,5 чел.-ч;
на холостые опыты - 2,0 чел.-ч;
на выполнение расчетов - 0,5 чел.-ч.
1. Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, с. 32 - 39.
3. Руководство по методам химического анализа морских вод. Л.: Гидрометеоиздат, 1977, с. 82 - 92, 101 - 109.
4. Руководство по химическому анализу поверхностных вод суши. Л.: Гидрометеоиздат, 1977, с. 306 - 308.
5. Methods of seawater analyses/Ed. by K.Grasshoff. Weinheim New York: Verlag Chemie, 1976, p. 168 - 173.
7. Strickland J.D.H., Parsons T.R. A practical handbook of sea-water analysis. Fish. Res. Board Canada, Bull. 167, 1968.
Раздел исключен с 1 июля 2014 года. - РД 52.10.779-2013, утв. Росгидрометом 25.09.2013.
Фенолы - высокотоксичные соединения, оказывающие крайне неблагоприятное воздействие на живой организм. В приоритетных списках загрязняющих природные воды веществ фенолы стоят на одном из первых мест, что объясняется большим объемом их мирового производства, а также высокой токсичностью.
Источниками поступления фенолов в морскую среду могут быть бытовые, промышленные и сельскохозяйственные сточные воды, аварийные разливы, утечки при транспортировке, а также перенос по воздуху в результате испарения с поверхности воды и почвы. Кроме того, в объектах морской среды присутствуют фенолы природного происхождения, продуцируемые морскими водорослями - макрофитами [6].
В последние годы значительное внимание уделяется анализу в природных водах хлор- и нитрозамещенных фенолов. Это обусловлено их высокой токсичностью и устойчивостью к метаболизму. Хлорфенолы попадают в воду в результате хлорирования сточной и питьевой воды, а также деградации пестицидов, с отходами целлюлозно-бумажного производства. Появление нитрофенолов в окружающей среде является следствием нефтехимического производства и деградации некоторых видов фосфорорганических пестицидов. Основные сведения об исследуемых фенолах приведены в табл. 22.
Таблица 22
Некоторые физико-химические и токсикологические
характеристики фенолов
Фенол | Брутто-формула (относительная молекулярная масса) | Температура <*>, °C | Растворимость в воде <**>, мг/л | ПДКв, мг/л | ПДКв.р., мг/л | |
плавления | кипения | |||||
Фенол | C6H6O (94) | 40,90 | 181,80 | 67000 40 | 0,001 | 0,001 |
2-Метилфенол | C7H8O (108) | 30,99 | 191,00 | 31000 | 0,05 | 0,003 |
2,5-Диметилфенол | C8H10O (122) | 74,85 | 211,13 | М.р. | 0,25 | Н.у. |
2,6-Диметилфенол | C8H10O (122) | 45,62 | 201,03 | М.р. | 0,25 | Н.у. |
3,4-Диметилфенол | C8H10O (122) | 65,11 | 226,94 | М.р. | 5,00 | Н.у. |
3,5-Диметилфенол | C8H10O (122) | 63,27 | 221,69 | М.р. | 5,0 | Н.у. |
3-Хлорфенол | ClC6H5O (129) | 32,80 | 214,00 | 26000 | Н.у. | Н.у. |
2,4-Дихлорфенол | Cl2C6H4O (163) | 45,00 | 210,00 | 460 | 0,002 | Н.у. |
2,4,6-Трихлорфенол | Cl3C6H3O (198) | 67,00 | 243,50 754 | 800 50 | 0,0004 | Н.у. |
2,3,4,5,6-Пентахлорфенол | Cl5C6HO (266) | 191,00 | 309,00 | 30,00 | 0,3 | Н.у. |
2-Нитрофенол | NC6H5O3 (139) | 45,00 | 216,00 | 2100 | 0,06 | Н.у. |
4-Нитрофенол | NC6H5O3 (139) | 114,00 | 279,00 (разл.) | 16000 | 0,02 | Н.у. |
Примечания: 1. М.р. - мало растворим; н.у. - не установлена. 2. ПДКв - предельно допустимая концентрация химического вещества в воде водоема; ПДКв.р. - предельно допустимая концентрация химического вещества в воде водоема, используемого для рыбохозяйственных целей. -------------------------------- | ||||||
Очень малые значения предельно допустимых концентраций фенолов в воде диктуют необходимость применения высокочувствительных и специфичных методов их определения.
Применяющаяся в практике химического мониторинга морской среды фотометрическая методика определения суммы фенолов и фенолоподобных веществ [3, 4] недостаточно чувствительна, не позволяет количественно анализировать индивидуальные фенолы и поэтому метрологически не аттестована. Указанная методика может быть использована для приближенных оценок фенольного загрязнения морской воды.
В настоящее Руководство включена высокочувствительная и специфичная газохроматографическая методика, которая дает возможность определять индивидуальные фенолы различных типов в морских и распресненных водах в диапазоне концентраций исследуемых веществ от 0,3 до 5000 мкг/л [2].
1.1. Сущность предлагаемого метода заключается в следующем: фенол и алкилфенолы анализируют в форме свободных фенолов на хроматографе с пламенно-ионизационным детектором (ПИД) с предварительным извлечением их из воды изобутилацетатом с добавкой высаливателя [1].
1.2. Хлор- и нитрофенолы определяют на приборе с детектором типа электронного захвата (ДЭЗ). Предварительно с целью оптимизации хроматографических условий анализа производят совмещенный с экстракцией перевод анализируемых веществ в ацетилпроизводные [5, 7].
1.3. Идентификацию осуществляют по времени удерживания в сравнении с контрольными образцами фенолов. Количественный расчет проводят методом соотнесения с градуировочными растворами фенолов по площадям пиков на хроматограммах. Показатели погрешности измерений рассчитаны в диапазонах концентрации алкилфенолов от 1 до 5000 мкг/л и хлор- и нитрофенолов от 0,3 до 160 мкг/л.
Мешающее влияние нейтральных веществ устраняют предварительно отмывкой подщелоченной водной пробы органическим растворителем, органических кислот - обработкой бикарбонатом натрия.
Для выполнения анализа применяются:
хроматограф любой марки с детекторами электронного захвата или постоянной скорости рекомбинации, или ионизационного резонанса и пламенно-ионизационным;
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104 (аналитические);
весы лабораторные 4-го класса точности с наибольшим пределом взвешивания 500 г по ГОСТ 24104 (технические);
сушильный шкаф по ГОСТ 13474;
насос водоструйный стеклянный по ГОСТ 25336 или пластмассовый КМ-1230 по ТУ 64-1-862;
плитка электрическая с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
баня водяная по ГОСТ 25336;
батометр типа ГР-15М по ТУ 25-04-1750 или пластмассовый 7-литровый, например системы ИОАН;
баллон газовый для азота по ГОСТ 949;
баллон газовый для водорода по ГОСТ 949;
компрессор для подачи воздуха к детектору хроматографа, например типа КВМ-8, или баллон со сжатым воздухом по ГОСТ 949;
редуктор кислородный по ГОСТ 6268;
редуктор водородный по ГОСТ 15150;
штатив лабораторный ШЛ с зажимом по ТУ 64-1-707;
шланги резиновые по ГОСТ 5496;
шланги вакуумные по ТУ 38-105881;
секундомер по ГОСТ 5072;
термометр ТЛ-5 1-А по ГОСТ 215;
микрошприц МШ-10М на 10 мкл по ТУ 25-05-2152;
колонки хроматографические стеклянные длиной 1,8 - 2,0 м с внутренним диаметром 3 мм по ТУ 25-05-2815;
колбы мерные 2-го класса точности на 100; 500 и 1000 мл по ГОСТ 1770;
колбы круглодонные на 500 мл по ГОСТ 25336;
колбы грушевидные на 150 мл по ГОСТ 25336;
пробирки мерные на 5 мл по ГОСТ 1770;
стаканы на 250 мл по ГОСТ 25336;
цилиндры мерные на 50 мл по ГОСТ 1770;
воронки химические типа В диаметром 50 - 80 мм по ГОСТ 25336;
воронки делительные на 50; 1000 и 2000 мл по ГОСТ 25336;
пипетки 2-го класса точности на 0,2; 2; 5 мл по ГОСТ 20292;
холодильник прямой по ГОСТ 25336;
аллонж, тип АИО, по ГОСТ 25336;
склянки стеклянные с притертой пробкой на 25; 30; 100 и 1000 мл (для экстрактов и проб воды);
фильтры бумажные, тип ФОМ, по ТУ 6-09-1678;
бумага индикаторная универсальная по ТУ 6-09-181;
инертон-супер зернением от 0,160 до 0,200 мм с нанесенной неподвижной жидкой фазой (НЖФ) OV-225 в количестве 5%;
инертон-супер зернением от 0,160 до 0,200 мм с нанесенной НЖФ SE-30 в количестве 5%;
инертон-супер зернением от 0,160 до 0,200 мм;
антифомсилан (АФС), 70%-ный раствор в эфире поли-трис-(диметил)-метил-оксиметилсилоксан по ФС 421865;
ацетон, ос.ч., по ТУ 6-09-3513;
гексан, ч., очищенный концентрированной серной кислотой и свежеперегнанный, по ТУ 6-09-3375;
изобутиловый эфир уксусной кислоты, очищенный раствором гидроксида натрия концентрацией 1,6 моль/л и свежеперегнанный, по ТУ 6-09-701;
уксусный ангидрид, ч.д.а., по ГОСТ 5815;
натрий сернокислый безводный, ч.д.а., по ГОСТ 4166;
натрий углекислый, х.ч., по ГОСТ 4201;
калий двухромовокислый, ч.д.а., по ГОСТ 4220;
натрия гидроксид, х.ч., по ГОСТ 4328;
серная кислота, х.ч., по ГОСТ 4204;
соляная кислота, х.ч., по ГОСТ 3118;
вода дистиллированная, очищенная изобутиловым эфиром уксусной кислоты, по ГОСТ 6709;
эфир этиловый технический перегнанный по ГОСТ 6265;
азот особой чистоты по ГОСТ 9239 или поверочный нулевой газ (ПНГ);
водород особой чистоты по ГОСТ 3022;
стандартный раствор фенола ГСО N 1762, концентрация 100 мкг/мл;
стандартный раствор 2-метилфенола ГСО N 1763, концентрация 500 мкг/мл;
препараты фенолов: 2,5-диметилфенол; 2,6-диметилфенол; 3,4-диметилфенол; 3,5-диметилфенол; 3-хлорфенол; 2,4-дихлорфенол; 2,4,6-трихлорфенол; 2,3,4,5,6-пентахлорфенол; 2-нитрофенол; 4-нитрофенол - стандартные образцы импортного (например, фирмы "Кемикэл сервис вестчестер", РА 19380, США) или отечественного производства с содержанием основного вещества не менее 99,5%.
Отбор проб осуществляют с помощью стеклянного или пластмассового батометра. Пробы воды объемом 1 л (для каждого вида анализа) без фильтрации немедленно переносят в стеклянные бутыли и закрывают притертыми пробками. Применение полиэтиленовой посуды, резиновых и полиэтиленовых пробок не допускается. Пробы воды хранят не более суток в темноте при комнатной температуре.
Гексановые и изобутилацетатные экстракты хранят в холодильнике в склянках с притертыми пробками при условии полного отсутствия в них воды. Срок хранения до 4 мес.
4.1.1. Безводный сульфат натрия для осушения экстрактов прокаливают в сушильном шкафу при температуре 200 - 250 °C в течение 7 - 8 ч. Прокаленный сульфат натрия хранят в герметически закупоренной склянке. Срок хранения не ограничен.
4.1.2. Навески сульфата натрия готовят взвешиванием 177 г безводного сульфата натрия.
4.1.3. Навески бикарбоната натрия готовят взвешиванием 10 г натрия углекислого кислого.
4.1.4. Хромовую смесь готовят перед употреблением растворением 9,9 г двухромовокислого калия в 100 мл концентрированной серной кислоты.
4.1.5. Раствор детергентов готовят растворением 10 г любого синтетического моющего средства в 1 л кипящей воды. Используют свежеприготовленный раствор.
4.1.6. 10%-ный раствор бикарбоната натрия готовят растворением 50 г бикарбоната натрия в 100 - 150 мл дистиллированной воды и последующим доведением объема раствора дистиллированной водой до 500 мл в мерной колбе. Срок хранения раствора 1 год.
4.1.7. Растворы гидроксида натрия концентрациями 1,6 и 4 моль/л готовят растворением 64 и 160 г щелочи в 150 - 200 мл дистиллированной воды и последующим доведением объемов растворов дистиллированной водой до 1 л в мерных колбах. Срок хранения растворов 1 год.
4.1.8. Гексан перед употреблением очищают следующим образом: 700 - 800 мл гексана помещают в делительную воронку на 1000 мл, добавляют 50 мл концентрированной серной кислоты и встряхивают в течение 5 мин. Жидкостям дают расслоиться, серную кислоту отбрасывают, затем повторяют встряхивание в аналогичных условиях еще раз с новой порцией серной кислоты. Гексан отмывают от остатков кислоты встряхиванием с дистиллированной водой (порциями по 100 мл) до нейтральной реакции промывных вод. Сушат безводным сульфатом натрия.
Очищенный от примесей гексан перегоняют с дефлегматором, отбрасывая первую порцию 75 - 80 мл, и собирают фракцию с температурой кипения 67 - 69 °C. Хранят гексан в стеклянной посуде с притертой пробкой не более 1 года.
4.1.9. Изобутиловый эфир уксусной кислоты перед использованием очищают следующим образом: 1000 мл реактива помещают в делительную воронку, добавляют 15 мл раствора гидроксида натрия (1,6 моль/л) и встряхивают в течение 3 мин, затем водно-щелочной слой отбрасывают, эфир промывают дистиллированной водой до нейтральной реакции промывных вод, сушат безводным сульфатом натрия и перегоняют, собирая фракцию с температурой кипения 116 - 117 °C. Хранят в темной стеклянной посуде не более 1 года.
Стеклянную посуду и стеклянную вату промывают в следующем порядке: горячим раствором детергентов, дистиллированной водой (трижды), хромовой смесью, дистиллированной водой (трижды), ацетоном. После промывания все оборудование и стеклянная вата сушатся при температуре 150 - 200 °C в течение 2 - 3 ч.
Пробу воды объемом 1 л помещают в делительную воронку и, добавляя по каплям раствор гидроксида натрия концентрацией 4 моль/л, доводят pH до 12 - 13. Приливают 40 мл н-гексана и энергично встряхивают в течение 3 мин. Дают слоям разделиться (10 - 12 мин). Нижний водный слой сливают в исходную емкость из-под пробы, гексановый экстракт отбрасывают. Пробу воды опять помещают в воронку и доводят pH до 6 - 7, приливая по каплям концентрированную соляную кислоту. Добавляют в пробу воды 10 г бикарбоната натрия и встряхивают до полного его растворения. Приливают 1 мл уксусного ангидрида, 30 мл гексана и энергично встряхивают воронку в течение 3 мин. Дают слоям разделиться (10 - 15 мин). Нижний водный слой сливают в исходную емкость из-под пробы, гексановый экстракт образовавшихся ацетилпроизводных фенолов сливают в склянку, пропуская через воронку с 20 - 25 г безводного сульфата натрия, ацетилирование и экстракцию повторяют еще раз с теми же объемами реагентов.
Водный слой сливают в емкость из-под пробы, гексановый же слой пропускают через ту же воронку с сульфатом натрия, присоединяя к первой порции гексана. Затем приливают к пробе 30 мл гексана и 0,5 мл уксусного ангидрида, встряхивают 3 мин, дают слоям разделиться, нижний водный слой отбрасывают, а гексановый присоединяют к предыдущим двум порциям, фильтруя через ту же воронку с сульфатом натрия. Емкость из-под пробы и делительную воронку обмывают гексаном дважды порциями по 3 мл и присоединяют смыв к экстракту, пропуская его через использованную для фильтрования экстракта воронку с сульфатом натрия. Допускается хранение экстракта в течение 4 мес.
Высушенный экстракт переносят в грушевидную колбу на 150 мл, обмывают склянку, в которой он хранился, порцией гексана 2 - 3 мл, присоединяют промывные порции гексана к экстракту и отгоняют гексан в вакууме водоструйного насоса в токе азота до объема около 3 мл на водяной бане при температуре не выше 35 °C. Применение смазки или других смазочных материалов не допускается. Концентрированный экстракт переносят в пробирку с помощью капилляра, обмывают колбу порцией гексана 1 - 2 мл, смыв гексана присоединяют к концентрату в пробирку тем же капилляром и помещают пробирку на водяную баню с температурой 30 - 40 °C, отдувая пары растворителя досуха инертным газом из баллона через редуктор. Растворяют сухой остаток 0,2 мл гексана и аликвоту (1 - 2 мкл) вводят в хроматограф с ДЭЗ.
В емкость с пробой воды объемом 1 л добавляют навеску 177 г сульфата натрия и встряхивают до насыщения, затем пробу воды помещают в делительную воронку на 2 л и, добавляя по каплям концентрированную соляную кислоту, доводят pH до 1 - 2 (контроль по индикаторной бумаге). Приливают 10 мл изобутилацетата и встряхивают 10 мин. Дают слоям разделиться (10 - 15 мин), водный слой сливают в исходную емкость из-под пробы, а органический - через воронку с 20 - 25 г безводного сульфата натрия переносят в склянки объемом 25 - 30 мл с притертой пробкой. Экстракцию повторяют еще раз с тем же объемом изобутилацетата. Экстракт помещают в ту же склянку. В таком виде экстракты можно хранить в холодильнике в течение 4 мес. Для дальнейшей обработки экстракт переносят в делительную воронку на 50 мл и встряхивают 3 мин с 1 мл 10%-ного раствора бикарбоната натрия. После отстаивания нижний водный слой отбрасывают, а в делительную воронку приливают 1 мл раствора гидроксида натрия концентрацией 4 моль/л и реэкстрагируют фенолы (5 мин). Реэкстракт переносят в пробирку с притертой пробкой и подкисляют концентрированной HCl до pH = 1...2, не давая смеси нагреться выше комнатной температуры. Затем в пробирку вносят 0,1 мл изобутилацетата, закрывают пробкой и встряхивают 5 мин. Из верхнего органического слоя микрошприцем отбирают аликвоту объемом 1 - 5 мкл для ввода в хроматограф.
1 - 2 мкл экстрактов (получение см. п. п. 4.3 - 4.5) вводят в испаритель хроматографа, снабженного ДЭЗ и ПИД соответственно, и записывают хроматограмму. После выхода пика ацетилпроизводного пентахлорфенола (при анализе хлор- и нитрофенолов) и пика 3,4-диметилфенола (при анализе алкилфенолов), имеющих наибольшие индексы удерживания, оставляют прибор в холостом режиме работы на 20 мин во избежание оседания на колонке возможных органических высокомолекулярных примесей, содержащихся в морской воде.
5.2.1. Холостое определение проводят перед анализом проб воды. Цель определения - проверка чистоты реактивов и материалов, используемых для анализа. Для выполнения холостого определения берут те же объемы реактивов, что и для одной пробы воды и проводят с ними последовательно все операции, описанные в п. п. 4.3 - 4.5.
5.2.2. Если время удерживания пиков на хроматограмме холостого опыта не совпадает ни с одним из анализируемых фенолов, то холостое определение повторяют для каждой партии реактивов.
5.2.3. Если же на хроматограмме холостого опыта имеются пики с временами удерживания, совпадающими с временами удерживания фенолов, то необходимо путем постадийного исследования установить, какой из реактивов загрязнен, и/или попытаться очистить его, или заменить этим же реактивом, но из другой партии. В случае, если загрязненной оказывается дистиллированная вода, необходимо провести ее очистку.
5.2.4. Для проверки чистоты используемой посуды ее ополаскивают порцией ацетона 3 мл и 1 мкл полученного смывного раствора вводят в испаритель хроматографа. Отсутствие на хроматограмме пиков (кроме пика, соответствующего растворителю) служит доказательством чистоты посуды.
5.2.5. Перед анализом каждой пробы проверяют чистоту микрошприца, используемого для ввода экстракта проб. Для этого набирают 1 мкл чистого ацетона и вводят в испаритель хроматографа. При появлении пиков на хроматограмме (кроме пика растворителя) дополнительно промывают шприц ацетоном и вновь проверяют на чистоту.
хлор- и нитрофенолов
6.1.1. Для приготовления стандартных растворов индивидуальных фенолов взвешивают на аналитических весах по 0,002 г 3-хлор-, 2,4-дихлор-, 2,4,6-трихлор- и пентахлорфенола. Переносят навески в отдельные мерные колбы вместимостью 100 мл, растворяют в небольшом количестве ацетона и доводят объем раствора до метки тем же растворителем. Каждому полученному раствору приписывают концентрацию 20 мкг/мл.
Для приготовления стандартных растворов 2-нитро- и 4-нитрофенолов взвешивают по 0,03 г фенолов и готовят растворы аналогично вышеописанным. Полученным растворам приписывают концентрации 300 мкг/мл. Растворы стабильны при хранении в холодильнике в течение 6 мес.
6.1.2. Непосредственно перед использованием готовят два стандартных ацетоновых раствора в мерных колбах вместимостью 100 мл. Раствор 1: отбирают 2 мл стандартного раствора 4-нитрофенола и по 1 мл стандартных растворов 3-хлор-, 2,4-дихлор- и пентахлорфенолов, доводят объем до метки. Раствор 1 содержит 6 мкг/мл 4-нитрофенола и по 0,2 мкг/мл 3-хлор-, 2,4-дихлор- и пентахлорфенола.
Раствор 2: отбирают 2 мл стандартного раствора 2-нитрофенола и 1 мл стандартного раствора 2,4,6-трихлорфенола. Доводят объем до метки. Раствор 2 содержит 6 мкг/мл 2-нитрофенола и 0,2 мкг/мл 2,4,6-трихлорфенола.
алкилфенолов
6.2.1. В качестве стандартных индивидуальных растворов фенола и 2-метилфенола используют их стандартные образцы с концентрацией 100 и 500 мкг/мл соответственно.
Для приготовления стандартных растворов индивидуальных алкилфенолов взвешивают на аналитических весах по 0,05 г 2,6-диметил-, 2,5-диметил-, 3,5-диметил-, 3,4-диметилфенола. Навески переносят количественно в мерные колбы вместимостью 100 мл, растворяют в небольшом количестве ацетона и доводят объем раствора до метки тем же растворителем. Каждый из полученных растворов имеет концентрацию 500 мкг/мл. Растворы стабильны при хранении в холодильнике в течение 6 мес.
6.2.2. Готовят стандартный раствор смеси алкилфенолов в ацетоне с содержанием 1 мкг/мл фенола и по 5 мкг/мл всех остальных алкилфенолов. Для этого в мерную колбу на 100 мл отбирают по 1 мл стандартных растворов собственно фенола и индивидуальных алкилфенолов и доводят объем раствора до метки ацетоном.
определения хлор- и нитрофенолов
6.3.1. Разделение всех определяемых хлор- и нитрофенолов в виде их ацетилпроизводных на насадочной колонке с одной и той же НЖФ не достигается, и необходимо применять две колонки с НЖФ разной полярности. В настоящей методике в качестве таких НЖФ выбраны OV-225 и SE-30. В выбранных условиях проведения анализа (см. п. 7.1.3) не разделяются ацетилпроизводные 2,4,6-трихлорфенола и 2-нитрофенола на колонке с НЖФ OV-225 и 2,4-дихлорфенола и 4-нитрофенола - с НЖФ SE-30.
6.3.2. Установление градуировочных характеристик всех хлор- и нитрофенолов проводят с использованием стандартных растворов 1 и 2 (см. п. 6.1.2). Для этого из раствора 1 в ряд мерных колб вместимостью 1 л отбирают 0,25; 0,5; 0,75; 1,0; 1,5; 2,0; 2,5 мл и доводят объемы дистиллированной водой до метки. Концентрации полученных растворов равны соответственно для каждого хлорфенола 0,05; 0,1; 0,15; 0,2; 0,3; 0,4; 0,5 мкг/л, а для 4-нитрофенола - 1,5; 3,0; 4,5; 6,0; 9,0; 12,0; 15,0 мкг/л. Эти растворы проводят через все стадии анализа (см. п. п. 4.3, 4.4). Аналитические растворы вводят в хроматограф с ДЭЗ, снабженный колонкой с OV-225.
Из раствора 2 в ряд мерных колб вместимостью 1 л отбирают 0,25; 0,5; 0,75; 1,0; 1,5; 2,0; 2,5 мл и доводят объемы дистиллированной водой до метки. Концентрации полученных растворов равны 0,05; 0,1; 0,15; 0,2; 0,3; 0,4; 0,5 мкг/л для 2,4,6-трихлорфенола и 1,5; 3,0; 4,5; 6,0; 9,0; 12,0; 15,0 мкг/л для 2-нитрофенола. Эти растворы проводят через все стадии анализа (см. п. п. 4.3, 4.4). Аналитические растворы вводят в хроматограф с ДЭЗ, снабженный колонкой с SE-30.
6.3.3. Градуировочные графики строят в координатах "площадь пика ацетилпроизводного хлор- или нитрофенола (см2) - концентрация соответствующего хлор- или нитрофенола (мкг/л)".
определения алкилфенолов
6.4.1. Для установления градуировочных характеристик в ряд мерных колб вместимостью 1 л вносят 1,0; 1,5; 2,5; 4,0; 6,0; 8,0; 10,0 мл стандартного раствора смеси алкилфенолов (приготовление см. п. 6.2.2) и доводят объем до метки дистиллированной водой. Полученные растворы имеют концентрации 1,0; 1,5; 2,5; 4,0; 6,0; 8,0; 10,0 мкг/л для собственно фенола и 5,0; 7,5; 12,5; 20,0; 30,0; 40,0; 50,0 мкг/л для всех алкилфенолов. Каждый градуировочный раствор проводят через все стадии анализа (см. п. 4.5). Аналитические растворы вводят в хроматограф с ПИД, снабженный колонкой с АФС.
6.4.2. Градуировочный график строится в координатах "площадь пика фенола (см2) - концентрация фенола (мкг/л)" для собственно фенола и "площадь пика соответствующего алкилфенола (см2) - концентрация алкилфенола (мкг/л)" для всех алкилфенолов.
6.4.3. В приведенных условиях выполнения измерений (см. п. 7.2.3) собственно фенол и 2-нитрофенол выходят одним пиком, и если при анализе проб морской воды на хроматографе с ДЭЗ, снабженном колонкой с SE-30, установлено присутствие 2-нитрофенола в анализируемой пробе воды, то пик 1 на хроматограмме (рис. 16) соответствует сумме собственно фенола и 2-нитрофенола. В таких случаях содержание фенола в пробе устанавливают по разности с учетом вычисленной концентрации 2-нитрофенола. Для этого дополнительно строят градуировочный график для фенола (ГГф) по хроматограммам градуировочной смеси алкилфенолов без 2-нитрофенола и градуировочный график для 2-нитрофенола (ГГнф) по хроматограммам градуировочной смеси без фенола.

длиной 2 м с неподвижной жидкой фазой антифомсилан (5%)
1 - фенол; 2 - 2-метилфенол; 3 - 2,6-диметилфенол;
4 - 2,5-диметилфенол; 5 - 3,5-диметилфенол;
6 - 3,4-диметилфенол.
хлор- и нитрофенолов
Стеклянные колонки длиной 1,8 - 2,0 м промывают смесью ацетона и диэтилового эфира, сушат и заполняют готовым носителем с нанесенной жидкой фазой следующим образом: один конец колонки закрывают тампоном из стеклянной ваты и подсоединяют к водоструйному насосу, а в другой конец через воронку засыпают приготовленный носитель небольшими порциями, постукивая колонку палочкой с резиновым концом, и следят за тем, чтобы носитель ложился равномерно. Заполненную колонку закрывают тампоном из стеклянной ваты, устанавливают в термостат хроматографа с ДЭЗ, не подсоединяя к детектору, и кондиционируют в токе азота (скорость 15 - 20 мл/мин) при температуре 100 - 120 °C в течение 4 - 6 ч и при температуре 180 - 200 °C в течение 7 - 8 ч.
алкилфенолов
Необходимый для заполнения стеклянной колонки длиной 1,8 - 2,0 м объем твердого носителя инертона-супер (около 10 г) взвешивают на технических весах и помещают в фарфоровую чашку. Навеску АФС (5% массы носителя - около 0,72 г) растворяют в диэтиловом эфире и добавляют к твердому носителю. Диэтиловый эфир испаряют на водяной бане при постоянном осторожном перемешивании до приобретения фазой воздушно-сухого состояния. Чашку с приготовленной насадкой помещают в сушильный шкаф и выдерживают при 100 °C в течение 1 ч. Далее заполняют стеклянную колонку длиной 1,8 м приготовленной насадкой и кондиционируют ее (см. п. 6.5).
хлор- и нитрофенолов
6.7.1. Подготовку хроматографа с ДЭЗ проводят в соответствии с инструкцией по эксплуатации. С помощью пенного расходомера устанавливают расход азота через колонку 23 - 26 мл/мин и на поддув детектора ДЭЗ 110 - 120 мл/мин. Оптимальный расход азота через колонку и на поддув детектора ДЭЗ определяется качеством получаемых хроматограмм. Подсоединяют колонку к детектору и проверяют герметичность соединений при помощи мыльной пены. Измеряют суммарный расход азота на выходе прибора. Эту величину контролируют ежедневно перед началом работ.
Устанавливают температуру термостата колонок 140 - 150 °C, температуру термостата детектора 220 - 230 °C, испарителя 210 - 220 °C, рабочий предел измерений на шкале электрометра 10 х 10-12 А, скорость диаграммной ленты 240 мм/ч.
6.7.2. Критерием полноты кондиционирования газохроматографической колонки является соответствие дрейфа и нерегулярных шумов нулевой линии паспортным данным прибора. После выхода прибора на режим для насыщения колонки вводят несколько раз по 1 мкл стандартного раствора смеси ацетилпроизводных хлор- и нитрофенолов (получение см. п. 6.3.2).
Определение параметров колонки и детектора проводят согласно Приложению 1.
6.8.1. Подготовку хроматографа с ПИД также проводят в соответствии с инструкцией по эксплуатации. Устанавливают расход азота через колонку 20 - 25 мл/мин, водорода - 25 - 30 мл/мин, воздуха - 280 - 300 мл/мин. Подсоединяют откондиционированную колонку к детектору и проверяют герметичность соединений при помощи мыльной пены. Устанавливают температуру термостата колонок 90 °C, термостата детектора - 230 - 240 °C, испарителя - 220 - 230 °C, рабочий предел измерений на шкале электрометра 2 х 10-12 А, скорость диаграммной ленты 180 мм/ч. Режим программирования температуры термостата колонок следующий: изотерма при 90 °C - 3 мин; линейное программирование 90 °C  200 °C со скоростью 10°/мин. (Критерий полноты кондиционирования колонки см. п. 6.7.)
200 °C со скоростью 10°/мин. (Критерий полноты кондиционирования колонки см. п. 6.7.)
6.8.2. Для насыщения колонки анализируемыми соединениями вводят 5 - 7 раз по 1 мкл стандартного раствора смеси алкилфенолов, после этого колонка готова к анализу.
Определение параметров колонки и детектора проводят согласно Приложению 1.
6.9.1. Для определения характеристик линейности диапазона детектирования дозируют стандартные смеси хлор- и нитрофенолов и стандартные смеси алкилфенолов шести концентраций, отличающихся не более чем в 2 раза, и проводят через все стадии анализа (см. п. п. 4.3 - 4.5). Вводят в испаритель хроматографа по 1 мкл полученных растворов и записывают хроматограммы на рабочей шкале электрометра. Рассчитывают площади пиков на полученных хроматограммах и определяют отношение (K) концентрации фенолов C к площадям пиков S:
K = C / S.
Линейность детектирования сохраняется для концентраций, при которых значения K отличаются не более чем на 5%:
 ,
,где индексы 1 и 2 относятся соответственно к предыдущей и последующей концентрациям стандартной смеси фенолов.
6.9.2. В случае отсутствия по какой-либо причине линейности детектирования следует построить градуировочный график для всех используемых рабочих шкал электрометра.
7.1.1. В испаритель хроматографа с ДЭЗ, снабженного колонкой с OV-225, вводят микрошприцем 1 мкл стандартного раствора 1 (получение см. п. 6.3.2) и записывают хроматограмму. Времена удерживания всех компонентов рассчитывают по трем результатам хроматографирования. Этот параметр необходимо проверять перед началом определений после выхода прибора на режим.
7.1.2. Затем вводят в испаритель 1 мкл экстракта пробы (см. п. п. 4.3, 4.4). Хлорфенолы и нитрофенол идентифицируют, сравнивая времена удерживания индивидуальных соединений на хроматограмме пробы морской воды с соответствующими пиками на хроматограмме стандартного раствора 1.
7.1.3. В случае идентификации на хроматограмме пробы воды, полученной на колонке с OV-225, пика с относительным временем удерживания 0,45 - 0,47, принадлежащем ацетилпроизводным 2,4,6-трихлор- и 2-нитрофенола, необходимо ввести в испаритель хроматографа с ДЭЗ, снабженного колонкой с SE-30, 1 мкл стандартного раствора 2 (см. п. 6.3.2), определить времена удерживания ацетилпроизводных 2,4,6-трихлорфенола и 2-нитрофенола, а затем ввести в испаритель 1 мкл экстракта этой пробы воды.
7.1.4. Условия хроматографирования ацетилпроизводных хлор- и нитрофенолов приведены в табл. 23.
Таблица 23
хлор- и нитрофенолов на НЖФ разной полярности
--------------------------------
<*> Условия приведены для газового хроматографа "Цвет-100", модель 110.
Параметр | НЖФ | |
OV-225 | SE-30 | |
1. Рабочие шкалы электрометра, A | 10·10-12 | 10·10-12 |
20·10-12 | 20·10-12 | |
50·10-12 | 50·10-12 | |
2. Скорость протяжки ленты, мм/ч | 240 | 240 |
3. Расход газов, см3/мин | ||
азота в колонку | 23 - 26 | 23 - 26 |
азота в детектор | 110 - 120 | 110 - 120 |
4. Температурный режим, °C | ||
колонки | 140 - 150 | 130 - 140 |
испарителя | 210 - 220 | 210 - 220 |
детектора | 220 - 230 | 220 - 230 |
7.1.5. Типические хроматограммы ацетилпроизводных хлор- и нитрофенолов представлены на рис. 17 и 18. Относительные времена удерживания по отношению к ацетилпроизводному пентахлорфенола даны в табл. 24.

Рис. 17. Хроматограмма смеси хлор- и нитрофенолов
(в виде ацетилпроизводных) на колонке длиной 2 м
с неподвижной жидкой фазой OV-225 (5%)
1 - 3-хлорфенол; 2 - 2,4-дихлорфенол;
3 - 2-нитрофенол + 2,4,6-трихлорфенол;
4 - 4-нитрофенол; 5 - 2,3,4,5,6-пентахлорфенол.
Рис. 18. Хроматограмма смеси 2-нитрофенола
и 2,4,6-трихлорфенола (в виде ацетилпроизводных)
1 - 3-хлорфенол; 2 - 2-нитрофенол;
3 - 2,4-дихлорфенол + 4-нитрофенол;
4 - 2,4,6-трихлорфенол; 5 - 2,3,4,5,6-пентахлорфенол.
Таблица 24
Относительные времена удерживания <*> ацетилпроизводных
хлор- и нитрофенолов (по ацетилпроизводному пентахлорфенола)
при хроматографировании на колонках с НЖФ разной
полярности <**>
--------------------------------
<*> Времена удерживания получены при анализе смеси хлор- и нитрофенолов на хроматографе "Цвет-100", модель 110.
<**> Параметры колонок приведены в табл. 23.
Хлор- и нитрофенолы | НЖФ | |
OV-225 | SE-30 | |
3-Хлорфенол | 0,15 - 0,17 | 0,09 - 0,11 |
2,4-Дихлорфенол | 0,19 - 0,21 | 0,19 - 0,21 |
2,4,6-Трихлорфенол | 0,45 - 0,47 | 0,40 - 0,42 |
2,3,4,5,6-Пентахлорфенол | 1,00 | 1,00 |
2-Нитрофенол | 0,45 - 0,47 | 0,12 - 0,13 |
4-Нитрофенол | 0,69 - 0,71 | 0,19 - 0,21 |
7.2.1. В испаритель хроматографа с ПИД вводят микрошприцем 1 - 5 мкл стандартного раствора алкилфенолов, включают кнопку программирования и записывают хроматограмму. Времена удерживания всех компонентов смеси рассчитывают по трем результатам хроматографирования. Этот параметр проверяют ежедневно перед началом определений после выхода прибора на режим.
7.2.2. Затем вводят в испаритель 1 - 5 мкл экстракта пробы (подготовку см. п. 4.5). Алкилфенолы идентифицируют, сравнивая времена удерживания компонентов пробы морской воды на полученной хроматограмме с соответствующими параметрами на хроматограмме смеси стандартных алкилфенолов.
Таблица 25
--------------------------------
<***> Условия приведены для газового хроматографа "Хром-5" (Чехо-Словакия).
Параметр | Значение |
Рабочая шкала электрометра, A | 2·10-12 |
4·10-12 | |
8·10-12 | |
16·10-12 | |
Скорость протяжки ленты, мм/ч | 180 |
Расход газов, см3/мин | |
азота | 20 - 25 |
водорода | 25 - 30 |
воздуха | 280 - 300 |
Температурный режим, °C колонки | Изотерма при 90 °C (3 мин) линейное программирование 90 - 200 °C со скоростью 10°/мин |
испарителя | 220 - 230 |
детектора | 230 - 240 |
7.2.4. Типичная хроматограмма смеси алкилфенолов приведена на рис. 16. Относительные времена удерживания по отношению к 3,4-диметилфенолу даны в табл. 26.
Таблица 26
Относительные времена удерживания <*> алкилфенолов
(по 3,4-диметилфенолу) при хроматографировании на АФС <**>
--------------------------------
<*> Времена удерживания получены при анализе на хроматографе "Хром-5" (Чехо-Словакия).
<**> Параметры колонки, см. табл. 25.
Алкилфенол | Относительное время удерживания |
Фенол | 0,25 - 0,27 |
2-Метилфенол | 0,31 - 0,33 |
2,6-Диметилфенол | 0,37 - 0,39 |
2,5-Диметилфенол | 0,64 - 0,66 |
3,5-Диметилфенол | 0,83 - 0,85 |
3,4-Диметилфенол | 1,00 |
8.1.1. Содержание фенолов в анализируемой пробе морской воды находят по формуле
 ,
,где  - концентрация соответствующего фенола в пробе, мкг/л;
- концентрация соответствующего фенола в пробе, мкг/л;
Площади пиков рассчитывают по формуле
 ,
,где S - площадь пика, см2;
h - высота пика, см;
8.1.2. Поскольку в пробе при анализе на хроматографе с ПИД в выбранных условиях фенол и 2-нитрофенол выходят одним пиком, вычисление концентрации фенола производят с учетом вычисленной ранее концентрации 2-нитрофенола (при анализе проб на хлор- и нитрофенолы на хроматографе с ДЭЗ). Для этого из суммарной площади двух фенолов вычитают площадь пика 2-нитрофенола, найденную по градуировочному графику ГГнф (см. п. 6.4). Затем по полученной величине площади пика фенола находят его концентрацию, используя ГГф.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09 по 20.12.89 (табл. 27), настоящая методика определения фенолов допущена к применению в организациях Росгидромета.
Таблица 27
Результаты метрологической аттестации МВИ
Фенол | Диапазон концентраций, мкг/л | Показатель воспроизводимости ( | Показатель правильности ( | Показатель погрешности МВИ, суммарная погрешность ( |
2-Нитрофенол | 18,5 - 30,7 | 5,0 | 20,0 | 20,0 |
2-Нитрофенол | 30,8 - 69,3 | 3,5 | 12,0 | 12,9 |
4-Нитрофенол | 2,0 - 6,3 | 3,6 | 8,5 | 9,5 |
6,4 - 12,7 | 2,9 | 7,9 | 8,7 | |
3,4-Диметилфенол | 60,0 - 120,0 | 3,0 | 8,4 | 9,2 |
120,1 - 5000 | 3,4 | 7,3 | 8,3 | |
3,5-Диметилфенол | 60,0 - 120,0 | 3,1 | 7,7 | 8,6 |
120,1 - 5000 | 3,3 | 7,2 | 8,2 | |
2,4-Дихлорфенол | 2,5 - 65,0 | 6,0 | 15,0 | 16,3 |
65,1 - 130 | 4,5 | 9,5 | 10,8 | |
2,4,6-Трихлорфенол | 0,3 - 2,5 | 5,0 | 16,6 | 17,9 |
2,6 - 16,3 | 4,6 | 10,6 | 12,4 | |
2,6-Диметилфенол | 30,0 - 60,0 | 2,4 | 7,6 | 8,3 |
60,1 - 120,0 | 1,9 | 5,2 | 5,7 | |
2,5-Диметилфенол | 60,0 - 120,0 | 2,9 | 6,9 | 7,7 |
120,1 - 250 | 2,3 | 6,8 | 7,5 | |
Фенол | 1,0 - 5,0 | 8,3 | 21,1 | 23,5 |
5,1 - 15,0 | 1,6 | 3,2 | 3,7 | |
2-Метилфенол | 6,0 - 15,0 | 4,3 | 9,2 | 10,5 |
15,1 - 30,0 | 1,7 | 5,8 | 6,3 | |
3-Хлорфенол | 13,5 - 27,0 | 5,7 | 11,5 | 13,2 |
27,1 - 83,0 | 3,3 | 8,3 | 9,3 | |
83,1 - 166,7 | 1,6 | 4,8 | 5,2 | |
2,3,4,5,6-Пентахлорфенол | 0,6 - 4,0 | 6,2 | 20,5 | 22,3 |
4,1 - 8,0 | 3,8 | 10,0 | 10,7 |
Анализ проб воды на содержание фенолов должен выполняться высококвалифицированными химиками-аналитиками, знакомыми с правилами эксплуатации приборов, применяемых в данной методике, и прошедшими соответствующий инструктаж по технике безопасности.
10.1. Для анализа 10 проб хлор- и нитрофенолов требуется 54,5 чел.-ч, в том числе:
на взятие проб из батометра - 0,5 чел.-ч;
на приготовление растворов реактивов - 6 чел.-ч;
на подготовку посуды - 4 чел.-ч;
на проведение пробоподготовки - 16 чел.-ч;
на подготовку прибора к измерениям - 4,0 чел.-ч;
на выполнение измерений - 20 чел.-ч;
на обработку значений, проведение расчетов, запись результатов - 4,0 чел.-ч.
10.2. Для анализа 10 проб алкилфенолов требуется 57,5 чел.-ч, в том числе:
на взятие проб из батометра - 0,5 чел.-ч;
на приготовление растворов и реактивов - 8 чел.-ч;
на подготовку посуды - 4 чел.-ч;
на проведение пробоподготовки - 22 чел.-ч;
на подготовку прибора к измерениям - 3,5 чел.-ч;
на выполнение измерений - 16 чел.-ч;
на обработку значений, проведение расчетов, запись результатов - 3,5 чел.-ч.
1. Коренман Я.И., Минасянц В.А., Фокин В.Н. Экстракционно-газохроматографическое определение микроколичеств фенолов в водных средах. Журнал аналитической химии, 1988, т. XIII, вып. 7, с. 1303 - 1306.
2. Методические указания. Определение фенолов в морской воде. РД 52.10.242-90. М.: Гидрометеоиздат, 1990, 44 с.
3. Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, с. 40 - 43.
5. Abrahamsson K., Xie T.M. Direct determination of trace amounts of chlorophenols in fresh water, waste and sea water. J. of chromatogr., 1983, 279, p. 199 - 208.
6. Buikema A., et al. Phenolics in aquatic ecosystems. Review. Mar. Environ. Res., 1979, N 2, p. 87 - 181.
7. Coutts A., Hargesheiner F., Passuto F. Application of a direct aqueous acetilation technique to the gas chromatographic quantitation of nitro-phenols and 1-naphtol in environmental water samples. J. of Chromatogr., 1980, 195, p. 105 - 112.
Хлорированные углеводороды (хлорорганические пестициды - ХОП и полихлорбифенилы - ПХБ) являются одними из наиболее опасных веществ, загрязняющих окружающую среду. Они попадают в морскую среду с промышленными и сельскохозяйственными стоками. Значительное количество этих соединений попадает в морскую среду из атмосферы.
Согласно данным комиссии по охране окружающей среды Балтийского моря [3], в середине 70-х годов во всем мире резко сократилось производство и применение ХОП и ПХБ. Содержание их в морской воде не превышает в настоящее время, как правило, десятых и сотых долей микрограмма в литре. Однако, являясь гидрофобными соединениями, хлорированные углеводороды преимущественно адсорбируются на взвешенных частицах, а также оседают на дно; кроме того, принимая во внимание их способность накапливаться в объектах морской среды (вода - донные отложения - планктон - рыбы - птицы), необходимо осуществлять контроль за фоновыми концентрациями этих соединений в морской воде (0,5 - 5,0 нг/л).
В основу настоящей методики положена работа Доусона и Райли [4].
Наиболее предпочтительным при определении ХОП и ПХБ в морской воде является метод газожидкостной хроматографии (ГЖХ) с использованием высокочувствительного к хлорорганическим соединениям детектора электронного захвата (ДЭЗ) [1].
Метод основан на извлечении хлорированных углеводородов органическим растворителем, очистке экстракта серной кислотой от мешающего влияния коэкстрагирующихся веществ и последующем детектировании ХОП в сконцентрированном экстракте на газовом хроматографе, снабженном детектором электронного захвата.
Идентификацию проводят по времени удерживания в сравнении с контрольными образцами ХОП и ПХБ. Количественный расчет проводят методом соотнесения с градуировочными растворами ХОП и ПХБ по высотам пиков на хроматограммах. Показатели погрешности измерений рассчитаны для  -ГХЦГ в диапазоне концентраций от 0,5 до 50,0 нг/л, для
-ГХЦГ в диапазоне концентраций от 0,5 до 50,0 нг/л, для  -ГХЦГ - от 0,4 до 20,0 нг/л, для ДДТ - от 3,0 до 200,0 нг/л, для ДДД - от 3,0 до 24,0 нг/л, для ДДЭ - от 2,0 до 150,0 нг/л (табл. 30).
-ГХЦГ - от 0,4 до 20,0 нг/л, для ДДТ - от 3,0 до 200,0 нг/л, для ДДД - от 3,0 до 24,0 нг/л, для ДДЭ - от 2,0 до 150,0 нг/л (табл. 30).
Для выполнения анализа применяют:
хроматограф любой марки, снабженный детектором типа электронного захвата или постоянной скорости рекомбинации;
колонки хроматографические стеклянные длиной 1,8 - 2,0 м с внутренним диаметром 3 мм по ТУ 25-05-2815;
микрошприц МШ-10м на 10 мкл по ТУ 25-05-2152;
эмалированное ведро;
батометр пластмассовый 7-литровый, например системы ИОАН;
баллон газовый для азота по ГОСТ 949;
редуктор кислородный по ГОСТ 6268;
штатив лабораторный ШЛ с зажимом по ТУ 64-1-707;
шланги резиновые по ГОСТ 5496;
шланги вакуумные по ТУ 38-105881;
секундомер по ГОСТ 5072;
термометр ТЛ-5 1-А по ГОСТ 215;
мешалка, снабженная электромотором, по ТУ 25-15-507;
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104 (аналитические);
сушильный шкаф по ГОСТ 13474;
центрифуга лабораторная любого типа, например ЦЛС-3, У-42, по ТУ 5.375-4170;
плитка электрическая с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
баня водяная по ТУ 46-22-608;
ротационный испаритель ИР-10 по ТУ 25-11-741;
прибор для отгонки растворителя, включающий в себя:
колбы круглодонные исполнения 1 на 500 мл по ГОСТ 25336;
холодильник прямой исполнения 1 по ГОСТ 25336;
аллонж типа АИО по ГОСТ 25336;
насос водоструйный стеклянный по ГОСТ 25336 или пластмассовый КМ-1230 по ТУ 64-1-862;
дефлегматор длиной 10 см по ГОСТ 20789;
бутыль для экстракции на 5 л по ГОСТ 10238;
встряхиватель типа АВУ по ТУ 64-1-1081;
колбы мерные 2 класса точности на 100 мл по ГОСТ 1770;
пипетки 2-го класса точности на 2 мл по ГОСТ 20292;
воронки делительные на 50 и 2000 мл по ГОСТ 25336;
пробирки мерные на 10 мл и 25 мл по ГОСТ 1770;
воронки Шотта по ГОСТ 9775;
колбы грушевидные на 300 мл по ГОСТ 25336;
воронки химические типа В диаметром 50 - 80 мм по ГОСТ 25336;
склянки стеклянные с притертой пробкой на 200 мл (для экстрактов) и на 5 л (для проб воды);
эксикатор по ГОСТ 6371;
бумага индикаторная по ТУ 6-09-1181;
фильтры бумажные типа ФОМ по ТУ 6-09-1678;
алюминиевая фольга;
стекловата;
хроматон N-AW-DMCS зернением от 0,160 до 0,200 мм с неподвижной жидкой фазой SE-30 в количестве 5% или DC-200 (3%);
гексан, ч., по ТУ 6-09-3375;
спирт этиловый ректификат высшей очистки по ГОСТ 5962 <*> или спирт этиловый ректификат высший сорт, перегнанный и пропущенный через активированный силикагель - по ГОСТ 18300;
--------------------------------
<*> На территории Российской Федерации действует ГОСТ Р 51652-2000, здесь и далее. - Примечание.
калия гидроксид, ос.ч. в гранулах - по ОСТ 6-01-301 или натрия гидроксид, х.ч. в гранулах - по ГОСТ 4328;
натрий сернокислый безводный, х.ч. - по ГОСТ 4171;
натрий углекислый кислый, х.ч. - по ГОСТ 4201;
серная кислота, х.ч. - по ГОСТ 4204;
кальций хлористый, ч. - по ТУ 6-09-4578;
силикагель L, хемапол (Чехо-Словакия), зернением 0,040 - 0,100 мм;
азот особой чистоты - по ГОСТ 9239 или поверочный нулевой газ (ПНГ);
ацетон, ос.ч. - по ТУ 6-09-3513;
бензол, х.ч. - по ГОСТ 5955;
синтетическое моющее средство любого типа;
стандартный раствор 4,4'-дихлордифенилдихлорэтена (ДДЭ) - ГСО N 4190, концентрация 100 мкг/мл;
стандартный раствор 4,4'-дихлордифенилтрихлорэтана (ДДТ) - ГСО N 4189, концентрация 100 мкг/мл;
препараты хлорированных углеводородов отечественного или импортного производства с содержанием основного вещества не менее 99,4%:
4,4'-дихлордифенилдихлорэтан (ДДД);
альфа-изомер гексахлорциклогексана;
гамма-изомер гексахлорциклогексана (линдан);
полихлорбифенилы - хлофен А-50.
Для отбора проб воды с горизонта 0 м при высоте волны до 1,5 м необходимо использовать узкогорлую бутыль с укрепленным на дне грузом. Если высота волны больше 1,5 м, для отбора проб с горизонта 0 м можно использовать батометр или ведро. С нижележащих горизонтов пробы отбирают стеклянным, металлическим или, в крайнем случае, пластмассовым батометром.
Консервации и хранению пробы не подлежат. В течение двух часов после отбора необходимо проэкстрагировать их н-гексаном с целью перевода ХОП и ПХБ в органическую фазу. Экстракты помещают в склянки с притертыми пробками, которые сверху дополнительно обертывают алюминиевой фольгой. Склянки помещают в ящики для экспедиционных грузов, прокладывают полиуретановыми прокладками и в таком виде транспортируют без дополнительного охлаждения.
Газохроматографический анализ экстрактов производят не позднее чем через 3 мес. после отбора пробы.
4.1.1. Безводный сульфат натрия для осушения экстрактов прокаливают в сушильном шкафу 6 - 8 ч при температуре 200 - 220 °C. Прокаленный сульфат натрия хранят в герметически закупоренной склянке. Срок хранения неограничен.
4.1.2. 1%-ный раствор бикарбоната натрия готовят растворением 2 г кристаллического бикарбоната натрия в 200 мл дистиллированной воды. Срок хранения раствора - 1 год.
4.1.3. Силикагель L прокаливают при температуре 320 - 350 °C в течение 25 - 30 ч. Хранят в эксикаторе с хлористым кальцием.
4.1.4. Кальций хлористый прокаливают при температуре 220 - 250 °C в течение 6 - 8 ч.
4.1.5. Гексан перед использованием перегоняют на приборе с дефлегматором, отбрасывая первую порцию в 75 - 80 мл, собирают фракцию с температурой кипения 67 - 69 °C и пропускают через колонку с 5 г активированного силикагеля.
4.1.6. Этиловый спирт перед использованием перегоняют на приборе с дефлегматором при температуре 78 °C и пропускают через колонку с 5 г силикагеля.
4.1.7. Раствор детергентов готовят растворением 10 г любого синтетического моющего средства в 1 л кипящей воды. Используют свежеприготовленный раствор.
Стеклянная и фарфоровая посуда промывается горячим раствором детергентов либо соды (на 1 л воды 10 г любого вещества), водопроводной водой, дистиллированной водой, ацетоном, гексаном. После промывания посуда сушится при температуре 130 - 140 °C в течение 4 ч и хранится завернутой в алюминиевую фольгу.
Из пробоотборника пробу морской воды объемом 4 л помещают в бутыль для экстракции объемом 5 л и экстрагируют 2 раза по 10 мин порциями по 100 мл перегнанного н-гексана. Экстракт отделяют в делительной воронке на 2 л, пропускают через стеклянный фильтр с 20 г безводного сульфата натрия в склянку для экстракта. Двумя порциями по 25 мл н-гексана обмывают делительную воронку и бутыль для экстракции, сливают в ту же склянку. Затем небольшими количествами (5 мл) н-гексана два раза промывают осушитель, слив присоединяют к экстракту.
Полученный экстракт упаривается до объема примерно 10 мл на ротационном испарителе или приборе для отгонки растворителя с вакуумом при температуре до 40 °C. Концентрат переливают в делительную воронку на 50 мл, добавляют 6 - 8 мл серной кислоты. Аккуратно переворачивают (но не встряхивают) воронку 15 - 20 раз, отбрасывают сернокислотный слой, а к органическому приливают еще 6 - 8 мл кислоты и переворачивают воронку 10 - 15 раз. Обработку серной кислотой проводят до тех пор, пока сернокислотный слой не будет оставаться бесцветным. Добавляют к очищенному экстракту 8 - 10 мл 1%-ного раствора бикарбоната, встряхивают, отбрасывают водный слой. Обработку бикарбонатом проводят до нейтральной реакции промывных вод по универсальной индикаторной бумаге. Органический слой пропускают через слой осушителя 5 г в мерную пробирку на 25 мл, осушитель промывают два раза н-гексаном порциями по 2 мл, слив присоединяют к экстракту.
Очищенный и осушенный экстракт упаривают до объема примерно 1 мл (объем записывают с точностью до 0,1 мл) под струей воздуха или при слабом нагревании пробирки на водяной бане при температуре не выше 40 °C.
--------------------------------
<*> Стадия дегидрохлорирования может быть опущена, если в анализе использовать капиллярные колонки, обладающие значительно лучшими параметрами разделения [2, 5]. Методика измерений с капиллярной колонкой полностью приведена в Приложении 2.
В случае одновременного присутствия в пробе морской воды ХОП и ПХБ эти группы необходимо разделить химически с помощью спиртового дегидрохлорирования. Для этого к концентрату (после того как из него отобраны 3 мкл и введены в хроматограф, который зафиксировал наличие ПХБ) приливают 1 мл этанола, добавляют одну гранулу гидроксида калия, закрывают пробирку стеклянной пробкой, тщательно размешивают и помещают в водяную баню при температуре 55 - 60 °C (не выше) на 30 мин. Затем пробирку охлаждают под струей водопроводной воды, добавляют к содержимому 3 мл дистиллированной воды, очень энергично встряхивают и дают отстояться в течение 5 мин до четкого разделения слоев (при возможности разделение слоев следует проводить в лабораторной центрифуге в течение 3 мин при 1500 об./мин). Из верхнего гексанового слоя отбирают аликвоту и вводят в хроматограф.
Отбирают 3 мкл экстракта из 1 мл концентрата (получение см. п. 4.4), вводят в испаритель хроматографа и записывают хроматограмму. После выхода пика ДДТ, имеющего наибольшее время удерживания, оставляют прибор в холостом режиме работы на 15 - 20 мин во избежание оседания на колонке органических высокомолекулярных примесей, которые могут содержаться в морской воде. В случае присутствия в пробе морской воды ПХБ, что выражается в появлении на хроматограмме большого числа пиков, по времени удерживания не совпадающих с пиками известных пестицидов, необходимо отобрать 3 мкл из верхнего гексанового слоя, полученного согласно п. 4.5, и ввести в хроматограф.
Перед тем как приступить к анализу проб морской воды, нужно проделать холостой опыт, чтобы убедиться в чистоте используемых реактивов. Для этого 200 мл перегнанного н-гексана концентрируют, обрабатывают серной кислотой, готовят к ГЖХ-определению (см. п. 4.4) и 3 мкл вводят в хроматограф. В дальнейшем такой холостой опыт нужно проводить с каждой новой партией гексана и кислоты.
При обнаружении загрязняющих веществ реактивы и посуду подвергают дополнительной очистке.
Перед анализом каждой пробы проверяют чистоту микрошприца, используемого для ввода экстракта проб. Для этого набирают 3 мкл чистого гексана и вводят в испаритель хроматографа. При появлении пиков на хроматограмме (кроме пика растворителя) дополнительно промывают шприц гексаном и вновь проверяют на чистоту.
Для приготовления стандартного раствора ХОП растворяют по 10 мг линдана и  -ГХЦГ и 80 мг ДДД в 100 мл перегнанного гексана, тщательно перемешивают, отбирают 0,5 мл полученной смеси, переносят в мерную колбу на 100 мл, добавляют туда же 2,5 мл ГСО ДДЭ и 5 мл ГСО ДДТ и доводят объем гексаном до метки. Отбирают из полученного раствора 1 мл, переносят в мерную пробирку на 10 мл и доводят объем гексаном до 10 мл.
-ГХЦГ и 80 мг ДДД в 100 мл перегнанного гексана, тщательно перемешивают, отбирают 0,5 мл полученной смеси, переносят в мерную колбу на 100 мл, добавляют туда же 2,5 мл ГСО ДДЭ и 5 мл ГСО ДДТ и доводят объем гексаном до метки. Отбирают из полученного раствора 1 мл, переносят в мерную пробирку на 10 мл и доводят объем гексаном до 10 мл.
Концентрации ХОП в растворе следующие: линдана - 50 нг/мл;  -ГХЦГ - 50 нг/мл; ДДЭ - 250 нг/мл; ДДД - 400 нг/мл; ДДТ - 500 нг/мл.
-ГХЦГ - 50 нг/мл; ДДЭ - 250 нг/мл; ДДД - 400 нг/мл; ДДТ - 500 нг/мл.
Для приготовления стандартного раствора ПХБ растворяют приблизительно 10 мг соединения хлофен А-50 в 100 мл гексана.
Полученный раствор тщательно перемешивают, отбирают 0,5 мл, переносят в мерную колбу на 100 мл, доводят объем гексаном до метки. Концентрация ПХБ в полученном растворе составляет приблизительно 500 нг/мл (рассчитывается с точностью до единиц нг/мл).
раствора ХОП и ПХБ
Для приготовления стандартного раствора смеси ХОП и ПХБ смешивают равные объемы стандартных растворов ХОП и ПХБ.
Концентрации отдельных компонентов ХОП и ПХБ в полученном растворе следующие: линдана - 25 нг/мл;  -ГХЦГ - 25 нг/мл; ДДЭ - 125 нг/мл; ДДД - 200 нг/мл; ДДТ - 250 нг/мл; ПХБ - 250 нг/мл.
-ГХЦГ - 25 нг/мл; ДДЭ - 125 нг/мл; ДДД - 200 нг/мл; ДДТ - 250 нг/мл; ПХБ - 250 нг/мл.
определения ХОП
В хроматограф при выбранных условиях (см. п. 7.4) вводят по 3 мкл стандартного раствора ХОП 3 - 5 раз. По результатам индивидуальных дозирований определяют среднюю высоту каждого пика и времена удерживания отдельных ХОП (рис. 19). Разбавляют раствор гексаном в 2, 4, 8 раз и записывают хроматограммы полученных растворов. Проводят математическую обработку полученных хроматограмм. Градуировочный график строят в координатах "высота пика - масса компонента". Убеждаются в его линейности для данного диапазона концентраций.

Рис. 19 - Хроматограмма смеси ХОП на колонке с SE-30
1 -  -гексахлорциклогексан; 2-
-гексахлорциклогексан; 2-  -гексахлорциклогексан
-гексахлорциклогексан
(линдан); 3 - ДДЭ; 4 - ДДД; 5- ДДТ
определения ПХБ
В испаритель хроматографа при выбранных условиях (см. п. 7.4) вводят несколько раз по 3 мкл стандартного раствора ПХБ - хлофена А-50. На полученной хроматограмме (рис. 20) определяют времена удерживания и среднюю высоту каждого из 14 самых высоких пиков отдельных компонентов хлофена А-50.

Рис. 20. Хроматограмма стандартной смеси ПХБ
(хлофен A-50 на SE-30)
Разбавляют стандартный раствор ПХБ в 2, 4, 8 раз и записывают хроматограммы полученных растворов. Проводят математическую обработку всех полученных хроматограмм. Градуировочный график строят в координатах "сумма высот самых интенсивных пиков ПХБ на хроматограммах стандартных растворов - масса хлофена А-50".
определения ХОП и ПХБ при их совместном присутствии
В испаритель хроматографа при выбранных условиях (см. п. 7.4) вводят несколько раз по 3 мкл стандартной смеси ХОП и ПХБ (приготовление см. п. 6.3). На полученной хроматограмме 1 (рис. 21) по результатам единичных вкалываний определяют среднюю высоту каждого пика. Затем берут 1 мл этой же смеси и проводят реакцию дегидрохлорирования (см. п. 4.5). Из верхнего гексанового слоя отбирают 3 мкл и вводят в хроматограф несколько раз при тех же условиях. Записывают хроматограмму 2 (рис. 22).

Рис. 21. Хроматограмма 1 на колонке с SE-30:
смесь ХОП и ПХБ до дегидрохлорирования

Рис. 22. Хроматограмма 2 на колонке с SE-30:
смесь ХОП и ПХБ после дегидрохлорирования
В результате дегидрохлорирования на хроматограмме 2 два пика (ДДТ и ДДД) исчезают, пик ДДЭ возрастает за счет ДДТ, появляется новый пик ДМЭ <*> (производное ДДД). Пики ПХБ при этом остаются прежней высоты.
--------------------------------
<*> ДМЭ - 4,4'-дихлордифенилхлорэтен.
Разбавляют смешанный стандартный раствор ХОП и ПХБ гексаном в 2, 4, 8 раз. Эти растворы также проводят через реакцию дегидрохлорирования и записывают их хроматограммы.
Градуировочный график в координатах "высота пика - концентрация ХОП (нг/мл)" для  -,
-, 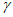 -ГХЦГ и ДДЭ строят, измеряя высоту пика на хроматограмме 1, полученной до дегидрохлорирования. Высота пика ДДТ подсчитывается как разность высот пиков, имеющих соответствующее время удерживания на хроматограммах 1 и 2 (т.е. до и после дегидрохлорирования). Аналогично рассчитывается высота пика ДДТ.
-ГХЦГ и ДДЭ строят, измеряя высоту пика на хроматограмме 1, полученной до дегидрохлорирования. Высота пика ДДТ подсчитывается как разность высот пиков, имеющих соответствующее время удерживания на хроматограммах 1 и 2 (т.е. до и после дегидрохлорирования). Аналогично рассчитывается высота пика ДДТ.
Для построения градуировочного графика ПХБ сравнивают высоты отдельных пиков ПХБ на хроматограммах 2 (после дегидрохлорирования) и стандартных растворов ПХБ. График строится в координатах "сумма высот отдельных пиков ПХБ - содержание ПХБ (нг/мл)".
Стеклянную колонку длиной 2 м (промытую последовательно бензолом и ацетоном) заполняют хроматоном N-AW-DMCS с НЖФ SE-30 (5%) или DC-200 (3%) следующим образом: закрывают один конец колонки стеклянной ватой, устанавливают марлевую прокладку и подсоединяют к водоструйному насосу, в другой конец колонки через воронку засыпают насадку; во время всей операции колонку аккуратно простукивают. После заполнения колонки закрывают свободный ее конец тампоном из стеклянной ваты. Колонку устанавливают в термостат колонок хроматографа, не подсоединяя к детектору электронного захвата, и кондиционируют в токе азота (30 мл/мин) при температуре 220 °C в течение 5 - 6 ч.
6.8.1. Подготовку хроматографа проводят в соответствии с инструкцией по эксплуатации. С помощью пенного расходомера устанавливают расход азота через колонку 25 - 30 мл/мин, через детектор 120 - 130 мл/мин. Оптимальный расход азота через колонку и на поддув детектора ДЭЗ определяется качеством получаемых хроматограмм. Подсоединяют колонку к детектору и проверяют герметичность соединений при помощи мыльной пены. Измеряют суммарный расход азота на выходе прибора. Эту величину контролируют ежедневно перед началом работ.
Устанавливают температуру термостата колонок 205 - 210 °C, детектора 215 - 220 °C, испарителя 225 °C, рабочий диапазон шкалы электрометра 10 х 10-12 - 50 х 10-12 А, скорость диаграммной ленты 240 мм/ч.
6.8.2. Критерием полноты кондиционирования газохроматографической колонки является соответствие дрейфа и нерегулярных шумов нулевой линии паспортным данным прибора. После выхода прибора на режим для насыщения колонки вводят несколько раз по 3 мкл стандартного раствора смеси ХОП.
Определение параметров колонки и детектора проводят согласно Приложению 1.
6.9.1. Для определения характеристик линейности диапазона детектирования готовят растворы стандартных смесей ХОП и ПХБ шести концентраций, различающихся не более чем в два раза. В испаритель хроматографа вводят по 3 мкл полученных растворов и записывают хроматограммы на рабочей шкале электрометра. Измеряют высоты пиков на полученных хроматограммах и определяют отношения концентраций С некоторых компонентов смеси ХОП и ПХБ к высотам пиков h:
К = С / h.
Линейность детектирования сохраняется для концентраций, при которых значения K отличаются не более чем на 5%:
 .
.6.9.2. В случае отсутствия по какой-либо причине линейности детектирования следует построить градуировочный график для всех используемых диапазонов.
7.1. В испаритель хроматографа вводят микрошприцем 3 мкл стандартного раствора ХОП и записывают хроматограмму. Времена удерживания всех компонентов рассчитывают по трем результатам хроматографирования. Этот параметр необходимо проверять перед началом определений после выхода прибора на режим.
Те же операции выполняют со стандартным раствором ПХБ и стандартной смесью ХОП и ПХБ.
7.2. Затем вводят в испаритель хроматографа 3 мкл экстракта пробы (подготовку см. п. п. 4.3 и 4.4). Отдельные компоненты ХОП идентифицируют, сравнивая времена удерживания индивидуальных соединений на хроматограмме пробы морской воды с соответствующими пиками на хроматограмме смеси стандартных веществ ХОП.
7.3. В том случае, если на хроматограмме экстракта пробы морской воды детектируются пики как ХОП, так и ПХБ, обрабатывают экстракт согласно п. 4.5 <*>, отбирают 3 мкл и вводят в хроматограф. На полученной хроматограмме пики идентифицируют согласно п. 6.6.
--------------------------------
<*> При использовании капиллярных колонок [2, 5], как указано выше, дегидрохлорирование не проводят.
Дегидрохлорирование следует проводить также и в том случае, если идентификация ХОП по одной хроматограмме не может быть проведена однозначно. Превращение ДДТ -> ДДЭ, ДДД -> ДМЭ является достаточно надежным критерием идентификации.
7.4. Условия хроматографирования ХОП и ПХБ на отечественном приборе "Цвет-100" мод. 110 приведены в табл. 28.
Таблица 28
Параметр | Значение |
Рабочие шкалы электрометра, А | 10 х 10-12 |
20 х 10-12 | |
50 х 10-12 | |
Скорость протяжки ленты, мм/ч | 240 |
Расход азота, см3/мин | |
в колонке | 25 - 30 |
в детекторе | 120 - 130 |
Температурный режим, °C | |
колонки | 205 - 210 |
испарителя | 225 |
детектора | 215 - 220 |
7.5. Типичные хроматограммы смеси ХОП и ПХБ представлены на рис. 21, 22. Относительные времена удерживания ХОП по отношению к ДДЭ приведены в табл. 29 (хроматограф "Цвет-100", мод. 110).
Таблица 29
Времена удерживания ХОП (относительно ДДЭ) на SE-30 <*>
--------------------------------
<*> Параметры колонки, см. табл. 28.
ХОП | Относительное время удерживания |
0,18 - 0,20 | |
0,25 - 0,27 | |
n,n'-ДДЭ | 1,00 |
n,n'-ДДД | 1,34 - 1,36 |
n,n'-ДДТ | 1,53 - 1,55 |
7.6. Времена удерживания ДДТ и ДДД на рекомендуемой колонке совпадают с временами удерживания отдельных пиков ПХБ, поэтому в случае их одновременного присутствия в пробах морской воды необходимо провести дегидрохлорирование (см. п. 4.5). В результате дегидрохлорирования пики ДДТ и ДДД исчезают, пик ДДД возрастает за счет ДДТ, появляется новый пик ДМЭ (за счет ДДД). Пики ПХБ при этом остаются прежней высоты.
в пробе только ХОП
Содержание ХОП в анализируемой пробе морской воды находят по формуле

где Cx - концентрация соответствующего ХОП в пробе, нг/л; Cст - концентрация соответствующего ХОП в стандартном растворе, нг/мл; hx - высота пика соответствующего ХОП на хроматограмме пробы морской воды, мм; hст - высота пика соответствующего ХОП в стандартном растворе, мм; Vэ - объем концентрата, проба которого берется для газохроматографического определения, мл; Vм.в. - объем пробы морской воды, взятой для анализа, л.
Расчет содержания ХОП производится по формуле (1) (см. п. 8.1), при этом высоты пиков  -,
-,  -ГХЦГ и ДДЭ измеряют по хроматограмме 1, полученной до дегидрохлорирования, так как мешающее влияние других соединений незначительно.
-ГХЦГ и ДДЭ измеряют по хроматограмме 1, полученной до дегидрохлорирования, так как мешающее влияние других соединений незначительно.
Высота пика ДДТ в исследуемом растворе подсчитывается как разность высот пиков, имеющих соответствующее время удерживания на хроматограммах 1 и 2 (т.е. до и после дегидрохлорирования).
Аналогично рассчитывается высота пика ДДД в исследуемом растворе.
Расчет ПХБ проводится по формуле
 ,
,где Cx, Cст, Vэ, Vм.в. - см. экспликацию к формуле (1);  - сумма высот пиков, соответствующих компонентам ПХБ в исследуемом растворе, мм;
- сумма высот пиков, соответствующих компонентам ПХБ в исследуемом растворе, мм;  - сумма высот пиков компонентов ПХБ в стандартном растворе хлофена А-50, мм.
- сумма высот пиков компонентов ПХБ в стандартном растворе хлофена А-50, мм.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09 по 20.12.90 (табл. 30), настоящая методика определения ХОП и ПХБ допущена к применению в организациях Росгидромета.
Таблица 30
Вещество | Диапазон концентраций, нг/л | Показатель воспроизводимости ( | Показатель правильности ( | Показатель погрешности МВИ, суммарная погрешность ( |
0,5 - 50,0 | 17,4 | 17,6 | 23,5 | |
0,4 - 20,0 | 14,6 | 2,0 | 14,6 | |
ДДТ | 3,0 - 200,0 | 11,2 | 10,0 | 14,4 |
ДДД | 3,0 - 24,0 | 7,5 | 4,8 | 8,4 |
ДДЭ | 2,0 - 150,0 | 15,0 | 19,6 | 21,6 |
Анализ проб воды на содержание ХОП и ПХБ должен выполняться высококвалифицированными химиками-аналитиками, знакомыми с правилами эксплуатации приборов, применяемых в данной методике, и прошедшими соответствующий инструктаж по технике безопасности.
Для анализа 10 проб ХОП и ПХБ требуется 65,5 чел.-ч, в том числе:
на взятие проб из батометра - 1,0 чел.-ч;
на приготовление реактивов и растворов - 4,0 чел.-ч;
на подготовку посуды - 3,0 чел.-ч;
на выведение прибора на режим - 4,0 чел.-ч;
на проведение экстракции, концентрирования экстрактов, ГЖ-анализа - 52,0 чел.-ч;
на проведение расчетов, запись результатов - 1,5 чел.-ч.
1. Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, с. 44 - 52.
2. Черняк С.М. Определение хлорированных углеводородов в объектах морской экосистемы. В кн.: Методологические основы комплексного глобального мониторинга океана. М.: Гидрометеоиздат, 1985, с. 41 - 56.
3. Baltic Sea Environment Proceedings, N 5 B, Assessement of the natural of the Baltic Sea, Helsinki Commission, 1981, 426 pp.
4. Dawson R., Riley J.P. Chlorine-containing pesticides and polychlorinated biphenyls in British coastal waters. Estuarine and Coastal Mar. Sci., 1977, N 4, p. 55 - 69.
5. Duinker J.C. Monitoring of cyclic organochlorins in the marine environment. Environ. Monitor. Assess., 1986, v. 7, p. 189 - 208.
Определение тяжелых металлов в морской воде является одной из основных задач мониторинга морской среды. Кадмий, свинец, медь, кобальт, никель, хром являются наиболее токсичными металлами, поступающими в морскую среду как при естественных процессах, так и в результате антропогенного воздействия. Железо и марганец, хотя и менее токсичны, играют важную роль в геохимическом поведении других токсичных тяжелых металлов, что необходимо учитывать при проведении мониторинга загрязнения морской среды.
В системе гидрометслужбы СНГ для определения металлов применяется спектрографический метод, разработанный в Гидрохимическом институте [4]. Однако этот метод характеризуется недостаточной чувствительностью и может быть использован только при определении высоких концентраций токсичных металлов, например, в шельфовых водах устьевых областей рек.
В последние годы для определения металлов широкое распространение получил метод атомно-абсорбционной спектрофотометрии [1, 2, 5, 6]. В то же время из-за мешающего влияния основного солевого состава морской воды этот метод позволяет проводить прямое определение лишь некоторых металлов - железа, марганца, хрома [3]. Для определения других элементов из числа вышеназванных необходимо предварительно произвести выделение их из морской воды. Чаще всего для выделения используют способ экстракции, который при проведении в мягких условиях позволяет определять лабильную, наиболее реакционноспособную форму токсичных металлов.
лабильных форм кадмия, свинца, меди, кобальта, никеля <*>
Раздел 1 исключен с 01.12.2021. - Приказ Росгидромета от 26.07.2021 N 246.
содержания тяжелых металлов в растворенном состоянии
Метод заключается в разложении металлорганических соединений кипячением пробы с соляной кислотой и персульфатом аммония с последующей нейтрализацией пробы, экстракции комплексов металлов с диэтилдитиокарбаматом натрия (НДДК) в тетрахлорметан, реэкстракции в кислый раствор и окончательном определении их содержания методом непламенной атомно-абсорбционной спектрофотометрии. Жесткая обработка проб воды окислителями в кислой среде особенно необходима при анализе распресненных и прибрежных вод, где содержание растворенных гуминовых веществ особенно высоко.
Настоящая методика может быть рекомендована для определения общего содержания тяжелых металлов в прибрежных распресненных водах.
Для выполнения анализа применяются:
атомно-абсорбционный спектрофотометр любой марки с непламенной атомизацией проб и дейтериевым корректором;
фильтровальное устройство типа ФМ-02 по ТУ 112.966.249;
электроплитка с закрытой спиралью ПЭК-800/3 по ТУ 92-208;
фильтры мембранные типа "Сынпор" диаметром 60 мм с размером пор 0,45 мкм;
колбы мерные на 0,1; 1 и 2 л по ГОСТ 1770;
воронки делительные на 0,5 л по ГОСТ 8613;
пипетки с делениями на 1; 2; 5 и 10 мл по ГОСТ 20292;
цилиндры мерные на 0,05; 0,25 и 0,5 л по ГОСТ 1770;
колба Бунзена на 1 л по ГОСТ 6514;
холодильники обратные с внутренним охлаждением со шлифом КШ 29 типа ХСВО-КШ по ГОСТ 9499;
колбы круглодонные со шлифом КШ 29 на 0,5 и 1 л по ГОСТ 10394;
флаконы полиэтиленовые на 50 мл по ТУ 6-19-45;
водоструйный стеклянный вакуумный насос по ГОСТ 25336 или насос вакуумный типа НВЭ по ТУ 79 РСФСР 102;
стандартные образцы для анализа вод СОВ-3, ГСО 1759;
N,N-диэтилдитиокарбамат натрия, ч.д.а., по ГОСТ 8864;
углерод четыреххлористый (тетрахлорметан), ос.ч., по ТУ 6-09-3219;
кислота азотная, ос.ч., по ГОСТ 11125;
кислота соляная, ос.ч., по ГОСТ 14261;
кислота уксусная, ос.ч., по ГОСТ 18270;
аммиак водный, ос.ч., по ТУ 6-09-19-91;
аммоний надсернокислый, х.ч., по ГОСТ 20478;
вода, ос.ч., по ТУ 6-09-2502;
азот газообразный, ос.ч., по МРТУ 6-02-375 или азот нулевой поверочный по ТУ 6-21-39.
2.3.1. Подготовка оборудования для отбора и фильтрования проб
Отбор проб производится согласно п. 1.3.1.
2.3.2. Фильтрация проб морской воды
Фильтрацию проб осуществляют согласно п. 1.3.2.
2.3.3. Консервация проб
Профильтрованные пробы подкисляют концентрированной соляной кислотой из расчета 10 мл кислоты на 1 л пробы. Соляная кислота в настоящей методике играет роль не только консерванта, но и участвует в последующем разрушении растворенного органического вещества в процессе кипячения с персульфатом аммония.
2.3.4. Хранение проб
Подкисленные пробы в полиэтиленовых флаконах упаковывают в полиэтиленовые мешки и хранят в неметаллических контейнерах. Подкисленные пробы можно хранить неограниченное время при условии полной герметичности флаконов.
2.4.1. Методы приготовления реактивов для проведения анализа
1. Ацетатный буферный раствор, pH = 4
Сливают вместе 480 мл концентрированной уксусной кислоты, 545 мл 25%-ного раствора аммиака и раствор разбавляют до 1,8 - 1,9 л деионизированной водой. Добавляют уксусную кислоту и устанавливают pH = 4 (контроль на pH-метре), после чего объем раствора доводят водой до 2 л в мерной колбе.
2. 2%-ный раствор диэтилдитиокарбамата натрия (НДДК)
Раствор диэтилдитиокарбамата натрия готовят согласно п. 1.4.1 (2).
2.4.2. Очистка ацетатного буферного раствора
Очистку ацетатного буферного раствора проводят следующим образом: к 0,5 л буферного раствора прибавляют 3 мл 2%-ного раствора НДДК, 20 мл четыреххлористого углерода и экстрагируют 3 мин. После отстаивания органический слой сливают. Экстракцию повторяют еще 2 - 3 раза с новыми порциями растворителя. Очищенный буферный раствор можно хранить в полиэтиленовой посуде с завинчивающейся крышкой длительное время.
2.4.3. Очистка раствора диэтилдитиокарбамата натрия
Очистку осуществляют согласно п. 1.4.2.
2.4.4. Подготовка посуды
Посуду очищают и моют согласно п. 1.4.3.
2.5.1. Схема проведения анализа
Профильтрованную пробу объемом 200 мл (500 мл для вод океанов и открытых районов морей) помещают в круглодонную термостойкую колбу объемом 500 мл с обратным холодильником, добавляют персульфат аммония из расчета 2 г персульфата аммония на 1 л пробы. Пробу в колбе нагревают до кипения и затем кипятят 30 мин. После охлаждения пробу переносят в делительную воронку емкостью 500 мл, раствором аммиака устанавливают pH ~= 4, добавляют 5 мл ацетатного буферного раствора, 3 мл 2%-ного раствора НДДК и 20 мл тетрахлорметана. Проводят экстракцию в течение 3 мин. После разделения слоев (через 5 - 10 мин) органический слой сливают в полиэтиленовый флакон. Повторяют экстракцию с 10 мл тетрахлорметана в течение 1 мин. Органические фазы объединяют, добавляют 0,2 мл концентрированной азотной кислоты и проводят реэкстракцию (30 с). Через 10 мин после реэкстракции к реэкстракту добавляют 4,8 мл деионизированной воды. Анализу подвергается верхний азотнокислый слой (реэкстракт) без его отделения или с отделением от органической фазы.
2.5.2. Холостое определение
Одновременно с обработкой проб проводят "холостой" опыт. Для этого 5 мл конц. соляной кислоты и 0,1 г персульфата аммония кипятят в колбе с обратным холодильником, охлаждают, нейтрализуют раствором аммиака до pH = 4, добавляют 5 мл ацетатного буфера, 3 мл 2%-ного раствора НДДК, 30 мл CCl4 и смесь энергично встряхивают 3 мин. После разделения органический слой сливают в полиэтиленовый флакон и проводят реэкстракцию (см. выше). Холостой опыт проводят не менее трех раз, а также каждый раз при замене одного или нескольких реактивов.
2.6.1. Методы приготовления градуировочных растворов
Основными стандартными растворами служат стандартные образцы для анализа вод СОВ-3, разработанные Физико-химическим институтом АН Украины и Екатеринбургским филиалом ВНИИМ им. Д.И. Менделеева.
Смешанный промежуточный стандартный раствор и рабочие градуировочные растворы готовят согласно п. 1.6.1.
2.6.2. Установление градуировочных характеристик метода
Приготовленные градуировочные растворы анализируют не менее трех раз на содержание каждого металла. Для определения кадмия, меди, свинца в графитовую кювету атомизатора вводят по 20 мкл соответствующих градуировочных растворов, а при определении кобальта и никеля - по 100 мкл. По полученным значениям абсорбции (среднее из трех значений) после вычитания абсорбции холостой пробы строят градуировочные графики в координатах "абсорбция - масса металла в 20 (100) мкл". Значения массы металлов в 20 (100) мкл представлены в табл. 34.
Таблица 34
Масса металлов в градуировочных растворах,
вводимых в графитовую кювету спектрофотометра
Металл | Объем градуировочного раствора, мкл | Масса металла, нг | |||||||
Кадмий | 20 | 0,02 | 0,10 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 | 2,00 |
Медь | 20 | 0,02 | 0,10 | 0,20 | 0,40 | 0,60 | 0,80 | 1,00 | 2,00 |
Свинец | 20 | 0,08 | 0,40 | 0,80 | 1,60 | 2,40 | 3,20 | 4,00 | 8,00 |
Кобальт | 100 | 0,10 | 0,50 | 1,00 | 2,00 | 3,00 | 4,00 | 5,00 | 10,00 |
Никель | 100 | 0,10 | 0,50 | 1,00 | 2,00 | 3,00 | 4,00 | 5,00 | 10,00 |
Анализ ведут в режиме абсорбции. Анализируемые реэкстракты вводят в графитовую кювету спектрофотометра с помощью микрошприцев Эппендорфа. Основные параметры атомно-абсорбционного анализа металлов в реэкстрактах с применением прибора "Перкин-Элмер", модель 503, см. п. 1.7.
При использовании других приборов оптимальные температурно-временные режимы анализа могут несколько отличаться.
2.8.1. Вычисление результатов измерений
По разности абсорбции анализируемой и холостой пробы находят по градуировочному графику массу металла (нг) в 20 (100) мкл реэкстракта. Расчет производится по формуле
 ,
,где Ci - концентрация металла в анализируемой пробе, мкг/л; C - масса металла по градуировочному графику, нг; Vр.э - объем реэкстракта, мл; Vал - объем впрыскиваемой аликвоты (0,02 или 0,1 мл); Vпр - объем проанализированной пробы, мл.
2.8.2. Числовые значения показателей погрешности
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 1 по 31 октября 1986 г. (табл. 35), настоящая методика определения общего содержания тяжелых металлов в растворенном состоянии в морской воде допущена к применению в организациях Росгидромета.
Таблица 35
Результаты метрологической аттестации
Определяемый металл | Диапазон концентраций тяжелых металлов в морской воде, мкг/л | Показатель воспроизводимости ( | Показатель правильности ( | Показатель погрешности МВИ, суммарная погрешность ( |
Медь | 3,6 - 5,6 | 1,46 | 17 | 17,3 |
Кадмий | 0,1 - 1,3 | 11,9 | 17 | 21,2 |
Свинец | 0,1 - 0,6 | 12,4 | 17 | 21,5 |
Никель | 1,1 - 2,7 | 2,39 | 17 | 17,5 |
Кобальт | 0,1 - 0,18 | 10,2 | 17 | 20,3 |
общего содержания растворенных железа, марганца и хрома
Раздел 3 исключен с 01.07.2014. - РД 52.10.778-2013, утв. Росгидрометом 25.09.2013.
Раздел 4 исключен с 01.07.2014. - РД 52.10.778-2013, утв. Росгидрометом 25.09.2013.
Раздел 5 исключен с 01.07.2014. - РД 52.10.778-2013, утв. Росгидрометом 25.09.2013.
1. Мамонтова С.А., Пчелинцева Н.Ф. Экстракционно-атомно-абсорбционное определение свинца и кадмия в природных водах, взвесях и осадках. Журнал аналитической химии, 1979, т. 34, N 11, с. 2231 - 2235.
2. Методические указания по определению токсичных загрязняющих веществ в морской воде на фоновом уровне, N 45. М.: Гидрометеоиздат, 1982, с. 5 - 11.
3. Определение общего содержания железа, марганца и хрома в растворенном состоянии в морской воде. Методические указания. РД 52.10.241-90. М.: Госкомгидромет, 1990, 11 с.
5. Сокольская Н.Н., Плетнева Т.И., Чупахин М.С. Аналитические характеристики атомно-абсорбционного спектрофотометра С-112 с непламенным атомизатором. Журнал аналитической химии, 1979, т. 34. N 4, с. 818 - 819.
6. Danielsson L.-G., Magnusson B., Westerlund S. An improved metal extraction procedure for the determination of trace metals in sea water by atomic absorption spectrometry with electrothermal atomization. Anal. Chim. Acta, 1978, v. 98, p. 47 - 57.
Методика позволяет определять общую растворенную ртуть в морских и распресненных водах и предназначена для проведения мониторинга этих вод. Она позволяет определять ртуть с пределом обнаружения 15 нг/л. Диапазон измеряемых концентраций общей растворенной ртути равен 15 - 150 нг/л. Анализу не мешают никакие ионы или соединения, обычно присутствующие в морской воде.
В морской воде растворенная ртуть всегда присутствует в различных физико-химических и химических формах [2]. Поэтому для нахождения ее общего содержания в морской воде все эти формы необходимо перевести в растворенную неорганическую (ионную) форму ртути, способы определения которой хорошо разработаны, в частности, методом непламенной атомной абсорбции. Обычно это превращение осуществляют в кислой среде с помощью сильных окислителей, таких как перманганат калия, персульфат калия, бром и др., в различных сочетаниях и режимах обработки [1, 3, 4].
Сущность методики заключается в том, что в профильтрованной через мембранный фильтр с размером пор 0,45 мкм и подкисленной пробе морской воды разрушают все растворенные формы ртути до ионов с помощью перманганата калия и персульфата калия при нагревании. Ионы ртути восстанавливают двухлористым оловом до металлической ртути, которую концентрируют выдуванием потоком очищенного воздуха в поглотительную ловушку с азотнокислым раствором перманганата калия, окисляющим металлическую ртуть. Ловушку подсоединяют к ртутному анализатору, удаляют окислитель гидроксиламином, а ионы ртути восстанавливают двухлористым оловом до металлической ртути. Ртутные пары выдувают в газовую кювету анализатора, где они поглощают УФ-излучение с длиной волны 253,7 нм, изменение интенсивности которого пропорционально концентрации ртути.
Для выполнения анализа применяются:
ртутный анализатор МАС-50 фирмы "Перкин-Элмер" (США);
батометр, например ГР-18, по ТУ 25-04-2507;
микрокомпрессор АЭН-1 "Скалярий" (для аквариумов) по ТУ 16-539, 630;
насос водоструйный стеклянный по ГОСТ 10696 или пластмассовый КМ-1230 по ТУ 64-1-861, или механический комовского типа НВК;
штатив лабораторный с зажимами и кольцами ШЛ по ТУ 64-1-707;
плитка электрическая бытовая ПЭК-800/3 по ТУ 92-208;
колбы Бунзена на 0,5 и 1,0 л по ТУ 25-11-1173;
воронка пластмассовая с крышкой типа воронки Бюхнера;
фильтры мембранные типа "Сынпор" (Чехо-Словакия) диаметром 60 мм и размером пор 0,45 мкм;
реакционный сосуд на 0,75 л (рис. 23);
из пробы морской воды
1 - скрубберы; 2 - поглотительная ловушка;
3 - реакционный сосуд; 4 - микрокомпрессор; 5 - шланги
поглотительная ловушка на 50 мл (см. рис. 23);
колбы мерные на 0,1; 0,5 и 1,0 л по ГОСТ 1770;
цилиндры мерные на 0,1; 0,5 и 1,0 л по ГОСТ 1770;
пипетки градуировочные на 1; 2; 5 и 10 мл по ГОСТ 20292;
пипетки на 5; 15 и 20 мл по ГОСТ 20292;
колбы плоскодонные на 0,5 л с НШ 29,0 по ГОСТ 10394;
стакан химический на 0,4 - 0,6 л по ГОСТ 10394;
склянки с притертыми пробками на 0,5 и 1,0 л;
колонки ионообменные (длина рабочей части 0,6 м, диаметр внутренний 4 см);
бутыль с притертой пробкой на 5 - 10 л;
бюксы низкие диаметром 80 мм по ГОСТ 7148;
стеклянный фильтр N 2;
стекла часовые;
трубки хлоркальциевые по ГОСТ 9964;
вата стеклянная;
пробки стеклянные с НШ 29,0 по ОСТ 25-79;
скрубберы пластмассовые (от ртутного анализатора);
трубка полихлорвиниловая (с внутренним диаметром 4 - 6 мм) по ТУ 64-1-2813;
сита лабораторные КСИ по ТУ 25-06-1250;
картон асбестовый по ГОСТ 2850;
пинцет пластмассовый;
кислота азотная конц., ос.ч., по ГОСТ 11125;
кислота соляная конц., ос.ч., по ГОСТ 14261;
натрий хлористый, ос.ч., по ТУ 6-09-3658;
натрия гидроксид, ос.ч., по ОСТ 6-01-302;
калий роданистый, х.ч., по ГОСТ 4139;
калий двухромовокислый (калий бихромат), х.ч., по ГОСТ 4220;
калий надсернокислый (калий персульфат), ч.д.а., по ГОСТ 4146;
калий марганцевокислый (калий перманганат), х.ч., по ГОСТ 20490;
смола ионообменная КУ-2-8 по ГОСТ 20298;
смола ионообменная АВ-17-2 (или ЭДЭ-10п) по ГОСТ 20301;
индикаторная бумага (универсальная pH = 1...10);
активный уголь БАУ по ГОСТ 6217.
Из перечисленных ниже реактивов для анализа необходимо применять только те, которые приложены к ртутному анализатору МАС-50:
основной стандартный раствор азотнокислой ртути, содержащий в 1 мл 1,0 мг ртути;
гидроксиламин солянокислый, 1,5%-ный раствор;
олово двухлористое, 10%-ный раствор.
Пробы морской воды отбирают пластмассовым батометром. Сразу же после отбора пробу необходимо профильтровать через очищенный мембранный фильтр с размером пор 0,45 мкм.
Если не предполагается немедленно после фильтрования анализировать пробу на ртуть, то фильтрат следует законсервировать, для чего его переливают в стеклянную емкость и добавляют 15 мл концентрированной азотной кислоты на каждые 0,5 л пробы и закрывают хорошо пришлифованной пробкой. При солености воды меньше 15+ в пробу дополнительно вносят 1 мл 5%-ного раствора бихромата калия.
Стеклянные емкости с законсервированными пробами следует хранить и перевозить в закрытых деревянных или пластмассовых ящиках.
4.1.1. Поглотительный раствор (0,1%-ный раствор перманганата калия в 5%-ной азотной кислоте) готовят разбавлением 10 мл 5%-ного раствора перманганата калия в 5%-ной азотной кислоте в мерной колбе на 500 мл с доведением раствора до метки. Его следует хранить в склянке из темного стекла в холодильнике. Он устойчив не более 3 сут.
4.1.2. Консервирующий реагент 1 готовят растворением 4 г бихромата калия в 1 л 5%-ной азотной кислоты. Он устойчив не менее месяца при хранении в темноте.
4.1.3. Консервирующий реагент 2 готовят разбавлением 5 мл консервирующего реагента 15%-ной азотной кислотой в мерной колбе на 100 мл до метки. Он устойчив не менее месяца при хранении в темноте.
4.1.4. Азотную кислоту, 5%-ный раствор, готовят смешением 38,4 мл концентрированной азотной кислоты и безртутной воды в мерной колбе на 500 мл с доведением раствора до метки.
4.1.5. Азотную кислоту, 1%-ный раствор, готовят смешением 7,7 мл концентрированной азотной кислоты и безртутной воды в мерной колбе на 500 мл с доведением раствора до метки.
4.1.6. Калий надсернокислый, 4%-ный раствор, готовят растворением 4 г соли в 96 мл безртутной воды.
4.1.7. Калий роданистый, 10%-ный раствор, готовят растворением 10 г соли в 90 мл безртутной воды.
4.1.8. Натрия гидроксид, 2%-ный и 4%-ный растворы, готовят растворением 2 и 4 г щелочи в 98 и 96 мл безртутной воды соответственно.
4.1.9. Соляную кислоту, раствор концентрацией 2 моль/л, готовят смешением 165 мл концентрированной соляной кислоты и безртутной воды в мерной колбе на 1000 мл с доведением раствора до метки.
В химический стакан на каждые 10 фильтров приливают 150 мл 1%-ной азотной кислоты. Закрывают его часовым стеклом, ставят на электроплитку, покрытую асбестовой тканью, и нагревают при слабом кипении раствора в течение часа. После охлаждения до комнатной температуры кислоту сливают. Очищенные фильтры можно хранить в течение нескольких недель в бюксах с хорошо притертыми крышками в свежей порции 1%-ной азотной кислоты.
Безртутную воду получают пропусканием дистиллированной воды через две последовательно соединенные колонки с ионообменными смолами КУ-2-8 и АВ-17-2 (или ЭДЭ-10п) со скоростью не более 60 капель в минуту. Система должна быть защищена от возможного попадания в безртутную воду загрязняющих веществ из воздуха лабораторного помещения, для чего на бутыли как с неочищенной дистиллированной водой, так и с очищенной безртутной водой надевают пробки, в которые вставлены хлоркальциевые трубки, наполненные активным углем.
Смолу катионообменную КУ-2-8 просеивают на ситах, отбирают фракцию с размером зерен 0,25 - 0,50 мм и выдерживают ее 20 ч в мерном цилиндре или химическом стакане, наполненном насыщенным раствором хлористого натрия. Этой смолой заполняют колонку и отмывают от пыли и осколков зерен пропусканием дистиллированной воды снизу вверх с такой скоростью, чтобы смола находилась во взвешенном состоянии. Отмывку прекращают при отсутствии в промывных водах взвешенных частиц. Затем смолу промывают дистиллированной водой сверху вниз до нейтральной реакции промывных вод по индикаторной бумаге.
Смолу анионообменную АВ-17-2 обрабатывают так же, как и катионит КУ-2-8, до заполнения ею колонки. Затем ее промывают 2%-ным раствором гидроксида натрия до бесцветной окраски вытекающего раствора, дистиллированной водой в объемном отношении к смоле 10:1 и соляной кислотой концентрацией 2 моль/л до исчезновения в вытекающем растворе ионов Fe3+, в присутствии которых при добавлении к 50 мл элюента 5 мл 10%-ного раствора роданида калия он окрашивается в красный цвет. Анионит промывают дистиллированной водой до нейтральной реакции промывных вод по индикаторной бумаге. Растворы пропускают через смолу снизу вверх. Анионит хранят в безртутной воде в склянке с притертой пробкой.
Смолу ЭДЭ-10п обрабатывают так же, как анионит АВ-17-2, только после удаления ионов железа соляной кислотой через нее пропускают 0,5 л 4%-ного раствора гидроокиси натрия на каждые 40 мл смолы для перевода ее в OH--форму. После этого анионит промывают дистиллированной водой до нейтральной После этого анионит промывают дистиллированной водой до нейтральной реакции промывных вод по индикаторной бумаге.
Подготовленными к работе смолами заполняют колонки слоем 45 - 50 см. Смолы необходимо регенерировать после пропускания через них 500 л дистиллированной воды.
Калий надсернокислый очищают двукратной перекристаллизацией из дистиллированной воды. Около 60 г соли растворяют при 70 °C в 250 мл воды и фильтруют горячий раствор (примерно 70 °C) через предварительно нагретый стеклянный фильтр N 2. По охлаждении фильтрата до 10 °C маточный раствор сливают. Операцию повторяют с осевшей на дне стакана солью и с таким же объемом воды, но без фильтрования. Охлажденный до 10 - 15 °C раствор фильтруют через стеклянный фильтр N 2, соль на фильтре тщательно отжимают стеклянной плоской пробкой. Окончательно соль высушивают в сушильном шкафу при 60 °C до постоянной массы. Перекристаллизованная соль не должна иметь запаха. Ее хранят в бюксе с притертой крышкой.
через пробу морской воды
Вставляют скруббер с активным углем в трубку между микрокомпрессором и реакционным сосудом; в последний наливают 575 мл дистиллированной воды, а отводную трубку опускают в цилиндр объемом 1 л, целиком заполненный водой и погруженный отверстием вниз в кастрюлю или таз с водой. Включают компрессор и сразу же секундомер (можно пользоваться часами с центральной секундной стрелкой). Пропускают воздух три минуты. С помощью регулятора скорости воздуха на микрокомпрессоре устанавливают ее в пределах 280 - 320 мл/мин.
Всю посуду сначала ополаскивают водопроводной водой, затем горячей хромовой смесью, снова водопроводной водой до полного удаления хромовой смеси, потом ополаскивают дистиллированной водой, азотной кислотой (1:1) и, наконец, безртутной водой.
Белый налет в реакционном сосуде и поглотительной ловушке от двухлористого олова удаляют щелочным раствором перманганата калия, а затем посуду отмывают так же, как от хромовой смеси.
Собирают прибор для концентрирования ртути из пробы морской воды, состоящей из реакционного сосуда (см. рис. 23), входное отверстие которого соединено с микрокомпрессором через скруббер с активным углем для очистки пропускаемого воздуха, а выходное отверстие - с поглотительной ловушкой, на выходном отверстии которой также надет скруббер с активным углем для защиты поглотительного раствора от загрязнения воздухом лаборатории. Активный уголь необходимо менять в скруббере для очистки пропускаемого воздуха через каждые 50 анализов, а в скруббере для защиты поглотительного раствора через каждые 100 анализов. Кроме того, в лабораторном помещении, где проводят определение ртути, следует полностью исключить работу с органическими растворителями и курение.
В плоскодонную колбу на 500 мл приливают из мерного цилиндра 500 мл морской воды, профильтрованной через мембранный фильтр с размером пор 0,45 мкм, а затем 15 мл концентрированной азотной кислоты и хорошо перемешивают. Если же проба ранее была подкислена, то кислоту вообще не добавляют. Приливают из пипетки 15 мл 5%-ного раствора перманганата калия и после перемешивания вращением колбы оставляют стоять на 15 мин, закрыв ее горло часовым стеклом. После этого добавляют из пипетки 15 мл 4%-ного раствора персульфата калия, перемешивают, закрывают колбу часовым стеклом, ставят ее на нагретую электроплитку, на которую положен асбестовый картон, и нагревают 2 ч. Затем колбу снимают с плитки и охлаждают до комнатной температуры. Удаляют избыток хлора в воздушном столбе над пробой в колбе продуванием воздуха, пропущенного через скруббер с активным углем, из микрокомпрессора в течение 5 мин. После продувки проба не должна пахнуть хлором. Добавляют 1,5%-ный раствор гидроксиламина до полного растворения осадка и обесцвечивания пробы, при этом колбу энергично встряхивают, закрыв ее пробкой. Обычно требуется 22 - 25 мл этого раствора.
В поглотительную ловушку вносят 20 мл поглотительного раствора, а в реакционный сосуд переливают обработанную окислителями пробу морской воды из колбы. Частично вставляют в сосуд аэратор, быстро приливают из пипетки 5 мл 10%-ного раствора двухлористого олова и тотчас же вставляют аэратор до конца. Включают микрокомпрессор и пропускают пузырьки воздуха через пробу со скоростью 280 - 320 мл/мин в течение 15 мин, после чего микрокомпрессор выключают. Отсоединяют реакционный сосуд и скруббер от поглотительной ловушки и подсоединяют ее к ртутному анализатору. Вынимают из ловушки аэратор и добавляют к находящемуся в ней поглотительному раствору 1 мл 1,5%-ного раствора гидроксиламина. После обесцвечивания раствора, которое происходит примерно через 40 с, добавляют к смеси 1 мл 10%-ного раствора двухлористого олова и немедленно вставляют аэратор до конца.
Для определения содержания ртути в реактивах вносят 15 мл концентрированной азотной кислоты в плоскодонную колбу на 500 мл, затем 15 мл 5%-ного раствора перманганата калия и через 15 мин - 15 мл 4%-ного раствора персульфата калия. Колбу покрывают часовым стеклом и ставят нагреваться на плитку в течение 2 ч. После охлаждения добавляют 22 - 25 мл 1,5%-ного раствора гидроксиламина до полного растворения осадка и обесцвечивания раствора.
Переносят его в реакционный сосуд, быстро приливают 5 мл 10%-ного раствора двухлористого олова. Определение повторяют еще раз. Измеряют светопропускание. Определение содержания ртути в реактивах следует проводить всякий раз при замене одного или нескольких реактивов.
Добавление перманганата калия и персульфата калия в колбу с пробой, ее нагревание на плитке, охлаждение, а также продувание воздушного столба колбы перед добавлением раствора гидроксиламина необходимо проводить только в вытяжном шкафу с хорошо действующей тягой, так как при окислении выделяется довольно много хлора. Воздух в лабораторном помещении, где проводятся измерения на ртутном анализаторе, должен быть чистым. Недопустимо присутствие в нем паров органических растворителей, особенно бензола и ацетона, а также хлора и табачного дыма.
Промежуточный стандартный раствор азотнокислой ртути готовят разбавлением 1 мл основного стандартного раствора азотнокислой ртути 5%-ной азотной кислотой с добавлением 50 мл консервирующего реагента 1 в мерной колбе на 1,0 л с доведением раствора до метки. 1 мл этого раствора содержит 1,0 мкг ртути. Раствор устойчив не менее 6 мес.
Градуировочные стандартные растворы азотнокислой ртути готовят следующим образом. В мерные колбы на 100 мл отбирают 0,5; 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 мл и т.д. промежуточного стандартного раствора ртути, добавляют по 5 мл консервирующего реагента 1 и разбавляют их 5%-ной азотной кислотой.
Уменьшение объема раствора, из которого должна определяться ртуть, от 100 до 20 мл вынудило отказаться от калибровки ртутного анализатора МАС-50 в соответствии с паспортом по шкале "XI" по одному реперу, так как на практике трудно снять показания на краю этой шкалы прибора, где лежат определяемые концентрации ртути. Поэтому для более точных расчетов необходимо строить градуировочный график по шкале светопропускания "Т%", которая имеет большую частоту делений по сравнению со шкалой "XI".
Для построения графика к анализатору подсоединяют вместо аэрационной склянки поглотительную ловушку (см. рис. 23). Аэратор вынимают из ловушки и кладут на чистую поверхность, а в ловушку вносят последовательно с помощью пипетки по 1 мл градуировочных стандартных растворов с содержанием ртути 5; 10; 20; 40; 60; 80; 100 нг и т.д., 19 мл поглотительного раствора и 1 мл 1,5%-ного раствора гидроксиламина. Раствор должен обесцветиться через 15 - 20 с после встряхивания ловушки. Затем аэратор частично вставляют в ловушку таким образом, чтобы дырочки находились на 1 см выше поверхности раствора. Придерживая его в таком положении, быстро приливают 1 мл 10%-ного раствора двухлористого олова и немедленно вставляют аэратор до конца. Записывают наибольшее отклонение стрелки прибора по шкале "Т%". Из трех значений светопропускания для каждой концентрации ртути рассчитывают их средние арифметические. Содержание ртути в реактивах определяют таким же образом, за исключением того, что в ловушку до приливания поглотительного раствора вносят 1 мл консервирующего реагента 2.
Из полученных средних арифметических значений Т% для каждой концентрации ртути рассчитывают оптическую плотность по формуле
 .
.Из полученного значения вычитают оптическую плотность содержащих ртуть реактивов и строят градуировочный график в координатах "D - содержание ртути (нг) в 20 мл поглотительного раствора" (что соответствует ее содержанию в 500 мл пробы), который должен быть прямолинейным и проходить через начало координат.
Градуировочный график необходимо проверять не реже 1 раза в месяц и каждый раз после того, как на ртутном анализаторе не работали более недели.
Для каждой пробы определяют наибольшее отклонение стрелки по шкале светопропускания "Т%" и записывают в журнал.
По измеренным значениям светопропускания с помощью градуировочного графика находят общее содержание растворенной ртути в морской воде и в применяемых в анализе реактивах, а также ее содержание только в реактивах. После вычитания второго значения из первого и умножения полученной разности на два получают концентрацию общей растворенной ртути в морской воде (нг/л).
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР (табл. 38), настоящая методика определения общей растворенной ртути допускается к применению в организациях Росгидромета.
Таблица 38
РЕЗУЛЬТАТЫ МЕТРОЛОГИЧЕСКОЙ АТТЕСТАЦИИ
Диапазон концентрации ртути, мкг/л | Показатель воспроизводимости | Показатель правильности | Показатель погрешности МВИ, суммарная погрешность |
0,016 - 0,120 | 10,0 | 20,0 | 25,0 |
Необходимая точность определения общей растворенной ртути может быть достигнута при правильном построении градуировочного графика, а также тщательном контроле чистоты посуды, реактивов и воздуха лабораторного помещения. Ртутный анализатор должен приходить в рабочее состояние не более чем через 45 - 50 мин после прогрева. В противном случае следует сменить помутневшие окошки в кювете прибора.
Определение может проводить квалифицированный химик-аналитик, имеющий среднее специальное или высшее образование, стаж работы не менее трех лет и знакомый с правилами эксплуатации анализатора ртути МАС-50.
Для анализа 10 проб требуется 30 чел.-ч, в том числе:
на взятие проб из батометра - 0,3 чел.-ч;
на подготовку посуды - 1 чел.-ч;
на приготовление растворов реактивов - 2,0 чел.-ч;
на подготовку 2,5 л очищенной воды - 2,0 чел.-ч;
на подготовку анализатора к работе - 1,0 чел.-ч;
на очистку мембранных фильтров - 0,5 чел.-ч;
на фильтрование проб - 3 чел.-ч;
на химическую обработку - 15 чел.-ч;
на проведение холостых определений - 4 чел.-ч;
на выполнение измерений - 0,5 чел.-ч;
на проведение расчетов и запись результатов - 0,7 чел.-ч.
1. Методические указания по химическому анализу распресненных вод морских устьевых областей рек и эпиконтинентальных морей, N 46. М.: Гидрометеоиздат, 1984, с. 64 - 73.
2. Прокофьев А.К. Химические формы ртути, кадмия и цинка в природных водных средах. Успехи химии, 1981, т. 50, N 1, с. 54 - 84.
3. Matsunaga K., Konishi S., Nishimura M. Possible errors caused prior to measuremen t of mercury in natural waters with special reference to seawater. Environ. Sci. Technol., 1979, vol. 13, N 1, p. 63 - 65.
4. Mercury Analysis Working Party of BITC. Standartisation of methods for the determination of traces mercury. Part 5. Determination of total mercury in water. Anal. Chim. Acta, 1979, vol. 109, p. 209 - 228.
Раздел исключен с 01.12.2021. - Приказ Росгидромета от 26.07.2021 N 246.
Симм-триазиновые гербициды широко распространены в сельском хозяйстве. Они применяются для борьбы с сорной растительностью на посевах кукурузы, хлопчатника, риса, лука, гороха, на виноградниках, а также для борьбы с растительностью в водоемах [1]. С водостоком гербициды могут попасть в ручьи и реки, а через них - в моря, в связи с чем возможно накопление их в устьевых и прибрежных зонах. Симм-триазиновые гербициды персистентны (сохраняют активность в почве в течение 2 - 3 лет после внесения), токсичны, а также обладают кумулятивными свойствами и способны накапливаться по трофической цепи.
Применение симм-триазиновых гербицидов, как и других представителей класса пестицидов, строго ограничено. В зависимости от токсичности каждого конкретного гербицида ПДК колеблется от 1 мкг/л до полного отсутствия. Основные сведения о симм-триазиновых гербицидах, применяемых на территории СНГ, приведены в табл. 41.
Таблица 41
Сведения о симм-триазиновых гербицидах,
применяемых в сельском хозяйстве на территории СНГ
Наименование гербицида | Синонимы | Название смесевого препарата, включающего гербициды | ПДК в рыбохозяйственных водоемах, мг/л | Растворимость в воде, мг/л | Токсикологическая характеристика |
Атразин | - | Агелон Майазин Нитразин | 0,005 | 33 | Малотоксичен |
Симазин | Тетразин Гезаприм | Ситрин | 0,0024 | 5,0 | То же |
Прометрин | Гезагард | Агелон Ситрин | 0,050 | 48,0 | " |
Пропазин | Приматол | - | Н/с | 8,6 | " |
Тербуметон | - | Карагард | Н/с | 130,0 | Среднетоксичен |
Семерон | Десметрин | - | 0,0005 | 580,0 | Малотоксичен |
Мезоранил | Азипротрин Бразоран | - | Н/с | 74,0 | Среднетоксичен |
Метазин | - | - | 1,0 | 10,0 | Малотоксичен |
Котофор | Дипротрин Санкап | 0,0003 | 16,0 | То же |
Примечание. Н/с - нет сведений.
Универсальным методом анализа симм-триазиновых гербицидов является метод газожидкостной хроматографии с термоионным детектором [3, 4]. Он селективен, информативен, доступен для массового применения. Предлагаемая методика позволяет определить сумму растворенных и взвешенных форм наиболее распространенных симм-триазинов в морских и распресненных водах указанным методом [2].
Настоящая методика предназначена для одновременного определения в морских и устьевых распресненных водах семи наиболее распространенных симм-триазиновых гербицидов: тербуметона, пропазина, атразина, симазина, семерона, мезоранила и метазина, а также прометрина, котофора и других.
Для выполнения анализа применяются:
хроматограф любой марки с термоионным детектором (с таблеткой CsBr);
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104;
весы лабораторные 4-го класса точности с наибольшим пределом взвешивания 500 г по ГОСТ 24104;
сушильный шкаф, например типа ШС-3;
ротационный испаритель, например типа ИР-1, ИР-1М;
насос водоструйный стеклянный по ГОСТ 25336 или пластмассовый КМ-1230 по ТУ 64-1-862;
плитка электрическая с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
баня водяная по ТУ 46-22-608;
батометр пластмассовый 7-литровый, например системы ИОАН;
баллон газовый для азота (гелия) по ГОСТ 949;
баллон газовый для водорода по ГОСТ 949;
компрессор для подачи воздуха к детектору хроматографа, например типа КВМ-8, или баллон со сжатым воздухом по ГОСТ 949;
редуктор кислородный по ГОСТ 6268;
редуктор водородный по ГОСТ 15150;
штатив лабораторный ШЛ с зажимами по ТУ 64-1-707;
шланги резиновые по ГОСТ 5496;
шланги вакуумные по ТУ 38-105881;
секундомер по ГОСТ 5072;
термометр ТЛ-5 1-А по ГОСТ 215;
микрошприц типа "Газохром-101" или МШ-1 по ТУ 25-05-2152;
колонки газохроматографические стеклянные длиной 1,5 - 2,0 м с внутренним диаметром 3 мм по ГОСТ 16285;
колбы мерные на 25; 50; 100 и 250 мл по ГОСТ 1770;
колбы круглодонные на 500 мл по ГОСТ 25336;
колбы грушевидные на 10 мл по ГОСТ 25336;
стаканы на 50; 250 и 400 мл по ГОСТ 25336;
цилиндры мерные на 50 мл по ГОСТ 1770;
воронки химические типа В диаметром 50 - 80 мм по ГОСТ 25336;
воронки делительные на 500 и 1000 мл по ГОСТ 25336;
пипетки на 0,2; 2,0; 5,0 по ГОСТ 20292;
пробирки на 10 мл по ГОСТ 1770;
холодильник прямой по ГОСТ 25336;
аллонж, тип АИО, по ГОСТ 25336;
фильтры бумажные, тип ФОМ, по ТУ 6-09-1678;
бумага индикаторная универсальная по ТУ 6-09-181;
склянки стеклянные с притертой пробкой на 300 мл (для экстрактов проб) и на 5000 мл (для проб воды);
капилляры стеклянные;
аппарат для перегонки растворителей;
хроматон N-AW-DMCS или N-AW-HMDS зернения 0,160 - 0,200 мм с нанесенной НЖФ карбовакс 20 М в количестве 5%;
хроматон N-AW-DMCS или N-AW-HMDS зернения 0,160 - 0,200 мм с нанесенной НЖФ SE-30 в количестве 5%;
вода дистиллированная по ГОСТ 6709;
ацетон, ос.ч., по ТУ 6-09-3513, или ч.д.а. по ГОСТ 2603;
метилен хлористый, х.ч., очищенный концентрированной серной кислотой и свежеперегнанный по ТУ 6-09-06-856, или хлороформ, х.ч., перегнанный по ТУ 6-09-06-800;
спирт этиловый ректификат высшей очистки по ГОСТ 5962 или спирт этиловый ректификат высший сорт, перегнанный по ГОСТ 18300;
натрий сернокислый безводный, ч.д.а., по ГОСТ 4166;
кислота серная (плотность 1,84), х.ч., по ГОСТ 4204;
калий двухромовокислый, ч.д.а., по ГОСТ 4220;
калия гидроокись, х.ч., по ГОСТ 24363;
азот особой чистоты по ГОСТ 9293 или поверочный нулевой газ (ПНГ);
водород особой чистоты по ГОСТ 3022;
стандартный раствор симазина - ГСО N 4196, концентрация 100 мкг/мл;
стандартный раствор атразина - ГСО N 4195, концентрация 100 мкг/мл;
стандартный раствор пропазина - ГСО N 4199, концентрация 100 мкг/мл;
гербициды тербуметон, прометрин, котофор, семерон, мезоранил, метазин - стандартные образцы импортного (например фирмы Jeigy, Швейцария) или отечественного производства с содержанием основного вещества не менее 99,5%.
Отбор проб морской воды осуществляется с помощью 7-литрового пластмассового батометра. Пробу без фильтрации немедленно переносят из батометра в стеклянные бутыли вместимостью 5 л и закрывают стеклянными пробками. Применение для хранения проб полиэтиленовой посуды, резиновых и полиэтиленовых пробок не допускается во избежание сорбции на них гербицидов.
Пробы воды хранят не более суток при комнатной температуре.
Экстракты в хлористом метилене (хлороформе) хранят в склянках с притертыми пробками в темноте при температуре 5 - 10 °С. Срок хранения до шести месяцев.
4.1.1. Безводный сульфат натрия для осушения экстрактов прокаливают в сушильном шкафу при температуре 250 - 300 °С в течение 7 - 8 ч. Прокаленный сульфат натрия хранят в герметически закупоренной склянке. Срок хранения неограничен.
4.1.2. Хромовая смесь готовится перед употреблением растворением 9,9 г двухромовокислого калия в 100 мл концентрированной серной кислоты.
4.1.3. Раствор детергентов готовят растворением 10 г любого синтетического моющего средства в 1 л кипящей воды. Используют свежеприготовленный раствор.
4.1.4. Раствор гидроксида калия концентрацией 4 моль/л готовят растворением 5,6 гидроксида калия в 10 - 15 мл дистиллированной воды и последующим доведением объема раствора до 25 мл в мерной колбе. Срок хранения раствора 1 год.
4.1.5. Хлористый метилен перед употреблением очищают следующим образом: 700 - 800 мл помещают в делительную воронку на 1000 мл, добавляют 50 мл концентрированной серной кислоты и встряхивают в течение 2 мин. Жидкостям дают расслоиться, сливают серную кислоту, отбрасывают ее, затем повторяют встряхивание в аналогичных условиях еще дважды с новыми порциями серной кислоты. Последняя порция кислоты после встряхивания должна быть бесцветной.
Хлористый метилен отмывают от остатков кислоты встряхиванием с дистиллированной водой (порциями по 150 мл) до нейтральной реакции промывных вод.
Очищенный от примесей таким образом хлористый метилен перегоняют с дефлегматором, отбрасывая первую порцию в 75 - 80 мл, и хранят в посуде из темного стекла не более 2 мес.
Хлороформ указанной марки перегоняют с дефлегматором без предварительной очистки серной кислотой. Хранят в посуде из темного стекла не более 3 мес.
Стеклянная посуда промывается в следующем порядке: горячим раствором детергентов, дистиллированной водой (три раза), хромовой смесью, дистиллированной водой (5 - 7 раз), ацетоном. После промывания посуда сушится при 150 - 200 °С в течение 2 - 3 ч.
Пробу воды объемом 5 л помещают в стеклянную бутыль и, добавляя по каплям раствор гидроксида калия концентрацией 4 моль/л, доводят до pH = 10. Перед каждой экстракцией в воду добавляют по 5 мл этилового спирта. Экстрагируют гербициды три раза по 2 мин порциями по 100 мл хлористого метилена вручную или мешалкой ГОИН.
После окончания экстракции в объединенные экстракты добавляют безводный сульфат натрия для осушения и выдерживают при комнатной температуре в вытяжном шкафу не менее суток.
Осушенные экстракты фильтруют небольшими порциями в круглодонную колбу вместимостью 500 мл и отгоняют хлористый метилен до сухого остатка, нагревая колбу на водяной бане с температурой не выше 40 °С при пониженном давлении (водоструйный насос) в токе азота или гелия. Применение вакуумной смазки или других смазочных материалов не допускается.
Сухой остаток в колбе смывают три раза порциями ацетона по 1 мл, раствор помещают в грушевидную колбу вместимостью 10 мл и отгоняют ацетон досуха в аналогичных изложенным выше условиях. Сухой остаток растворяют в 0,5 мл ацетона. Полученный раствор используют для анализа на хроматографе. Допускается хранение экстракта в течение 1 мес. при температуре от 0 до 4 °С.
1 мкл экстракта, полученного согласно п. 4.4, вводят в испаритель хроматографа и записывают хроматограмму. После выхода метазина, имеющего наибольший индекс удерживания, прибор оставляют в холостом режиме работы на 30 мин во избежание возможного оседания в колонке органических высокомолекулярных примесей, содержащихся в морской воде.
Холостое определение проводят перед анализом проб воды. Цель определения - проверить чистоту реактивов и материалов, используемых для анализа. Для выполнения холостого определения берут тот же объем экстрагента (хлористого метилена, хлороформа), этилового спирта и раствора гидроксида калия концентрацией 4 моль/л, что и для анализа одной пробы воды, и проводят последовательно с ними все операции, описанные в п. п. 4.4 и 4.3.
При отсутствии пиков на хроматограмме холостого опыта холостое определение повторяют для каждой новой партии реактивов.
Если на хроматограмме холостого опыта появляются пики с временами удерживания, близкими к временам удерживания исследуемых веществ, то необходимо путем постадийного исследования установить, какой из реактивов загрязнен, и заменить его таким же реактивом из другой партии.
Для проверки чистоты используемой посуды ее ополаскивают 3 мл ацетона и 1 мкл полученного смывного раствора вводят в испаритель хроматографа. Отсутствие на хроматограмме пиков (кроме пика, соответствующего растворителю) служит доказательством чистоты посуды.
Особое внимание следует обратить на чистоту шприца, используемого для дозирования экстрактов проб. Для этого перед анализом каждой пробы набирают в шприц 1 мкл чистого ацетона и вводят в испаритель хроматографа. При появлении пиков на хроматограмме (кроме пика растворителя) следует дополнительно промыть шприц ацетоном и вновь проверить на чистоту.
6.1.1. Приготовление растворов симазина, атразина и пропазина
Для приготовления стандартных растворов симазина, атразина и пропазина вскрывают ампулы, содержащие по 1 мл раствора образца с концентрацией 0,1 мг/л, быстро переносят их содержимое в мерную колбу с притертой пробкой вместимостью 50 мл и доводят ацетоном до метки. Тщательно перемешивают содержимое колбы. Полученные растворы имеют концентрацию 20 мкг/мл.
Рабочие растворы симазина, атразина и пропазина готовят разбавлением основных стандартных растворов в 10 раз. Для этого 2,5 мл соответствующего раствора переносят в мерную колбу с притертой пробкой вместимостью 25 мл и доводят ацетоном до метки. Полученные растворы имеют концентрацию вещества 2 мкг/мл.
6.1.2. Приготовление растворов тербуметона, прометрина, семерона, котофора, мезоранила и метазина
Для приготовления стандартных растворов перечисленных гербицидов взвешивают последовательно по 10 мг тербуметона, прометрина, семерона, котофора и по 20 мг мезоранила и метазина. Навески помещают в мерные колбы на 50 мл, растворяют в небольшом количестве ацетона, а затем доводят до метки тем же растворителем. Полученные растворы тербуметона, прометрина, семерона и котофора имеют концентрацию 200 мкг/мл, растворы мезоранила и метазина - 400 мкг/мл.
Рабочие растворы этих веществ готовят разбавлением основных в 100 раз. Для этого отбирают по 0,5 мл каждого стандартного раствора, переносят в мерные колбы на 50 мл и доводят до метки ацетоном. Полученные таким образом рабочие растворы тербуметона, прометрина, семерона и котофора имеют концентрацию 2 мкг/мл, а мезоранила и метазина - 4 мкг/мл.
6.1.3. Приготовление раствора смеси гербицидов
Готовят стандартный раствор смеси гербицидов в ацетоне с содержанием тербуметона, атразина, пропазина, симазина, семерона по 1,0 мкг/мл; мезоранила и метазина по 2,0 мкг/мл. Для этого в мерную колбу емкостью 50 мл пипеткой переносят по 2,5 мл стандартных растворов атразина, пропазина и симазина и по 0,25 мл стандартных растворов тербуметона, семерона, мезоранила и метазина. Содержимое колбы перемешивают и доводят ацетоном до метки.
Растворы стандартных веществ стабильны при хранении в холодильнике в течение 6 мес.
В хроматограф при условиях, выбранных согласно п. 6.4, вводят по 1 мкл индивидуальных рабочих стандартных растворов гербицидов и определяют их времена удерживания. Затем записывают хроматограмму 1 мкл стандартного раствора смеси гербицидов. Разбавляют раствор в 2; 4; 8 раз и также записывают хроматограммы полученных растворов. Проводят математическую обработку всех полученных хроматограмм и, построив график зависимости площади пика от количества вещества для каждого компонента, убеждаются в его линейности для данного диапазона концентраций.
Стеклянную колонку длиной 1,5 - 2,0 м и диаметром 3 мм промывают ацетоном, сушат и заполняют готовой насадкой. Для этого один конец колонки плотно закрывают небольшим тампоном из стекловаты, промытой ацетоном, затем кусочком марли и подсоединяют к водоструйному насосу. После этого через свободный конец заполняют колонку насадкой, засыпая ее небольшими порциями и уплотняя постукиванием по колонке палочкой с резиновым наконечником при постоянно работающем насосе. Необходимо следить за тем, чтобы носитель ложился равномерно, без пустот, с одинаковой плотностью. Заполненную колонку закрывают тампоном из стекловаты и помещают в термостат колонок хроматографа, не подсоединяя к детектору. Кондиционируют колонку в токе газа-носителя при расходе его 15 - 20 мл/мин при температуре 100 °С в течение 7 - 8 ч и при температуре 200 °С в течение 5 - 6 ч.
Подготовку хроматографа проводят в соответствии с инструкцией по эксплуатации.
Устанавливают с помощью пенного расходомера расход азота (гелия), например 35 - 36 мл/мин. Подсоединяют колонку к детектору и проверяют герметичность соединений, нанося на них кисточкой мыльную пену.
Устанавливают поступление в детектор водорода со скоростью 12 - 14 мл/мин и воздуха 240 - 260 мл/мин.
Устанавливают температуру термостата колонок 190 - 200 °С, испарителя 200 - 220 °С, термостата детектора 220 - 240 °С. Поджигают водород в детекторе, устанавливают рабочий диапазон измерений на шкале электрометра и скорость диаграммной ленты (0,6 см/мин). При работе в рабочем диапазоне измерений электрометра высота пика пропазина, соответствующая 1 мкл стандартного раствора с концентрацией 1 мкг/мл, должна составить около 1/3 шкалы самописца.
Критерием полноты кондиционирования хроматографической колонки является отсутствие дрейфа и нерегулярных шумов нулевой линии.
Для насыщения колонки исследуемыми соединениями вкалывают подряд 10 - 12 раз по 1 мкл стандартного раствора пропазина с концентрацией 10 мкг/мл, после чего колонка полностью готова к работе.
Определение параметров колонки и детектора проводят согласно Приложению 1.
Для определения характеристик линейности диапазона детектирования готовят растворы стандартной смеси гербицидов шести концентраций, различающихся не более чем в два раза. По 1 мкл полученных растворов вводят в испаритель хроматографа и записывают хроматограммы на рабочей шкале электрометра. Рассчитывают площади пиков на полученных хроматограммах и определяют отношение концентрации гербицидов к площади пиков: (К = С / S).
Линейность детектирования сохраняется для концентраций, при которых значения К отличаются не более чем на 5%:
 .
.В случае отсутствия по какой-либо причине линейности детектирования следует построить градуировочный график для всех используемых диапазонов чувствительности.
В испаритель хроматографа вводят микрошприцем 1 мкл стандартного раствора смеси гербицидов и записывают хроматограмму. Времена удерживания всех компонентов рассчитывают по трем результатам хроматографирования. Этот параметр необходимо проверять ежедневно перед началом определений после выхода прибора на режим.
Затем вводят в испаритель 1 мкл экстракта пробы, подготовленного, как указано в п. п. 4.3 и 4.4. Симм-триазиновые гербициды идентифицируют, сравнивая времена удерживания индивидуальных соединений на хроматограмме пробы морской воды с соответствующими пиками на хроматограмме смеси стандартных веществ.
Условия хроматографирования смеси симм-триазиновых гербицидов приведены в табл. 42.
Таблица 42
гербицидов на колонках с НЖФ различной полярности
--------------------------------
<*> Условия приведены для газового хроматографа "Хром-5" (Чехо-Словакия).
Параметр | Колонка | |
размеры 1,5 м х 3,0 мм; карбовакс 20 М (5%) на хроматоне зернения 0,16 - 0,20 мм (I) | размеры 2,0 м х 3,0 мм; SE-30 (5%) на хроматоне зернения 0,16 - 0,20 мм (II) | |
1. Рабочая шкала электрометра, А | 4·10-12 | 4·10-12 |
8·10-12 | 8·10-12 | |
16·10-12 | 16·10-12 | |
2. Скорость протяжки ленты, мм/мин | 6 | 6 |
3. Расход газов, см3/мин | ||
азота (гелия) | 32 - 38 | 32 - 38 |
водорода | 12 - 14 | 12 - 14 |
воздуха | 240 - 260 | 240 - 260 |
4. Температурный режим, °С | ||
колонки | 190 - 200 | 180 - 190 |
испарителя | 205 - 215 | 200 - 210 |
детектора | 220 - 230 | 215 - 225 |
Типичные хроматограммы смеси симм-триазиновых гербицидов представлены на рис. 24 и 25. Времена удерживания относительно пропазина приведены в табл. 43.

Рис. 24. Хроматограмма смеси симм-триазиновых гербицидов
на колонке длиной 1,5 м с НЖФ карбоваксом 20 М (5%)
1 - тербуметон; 2 - пропазин; 3 - атразин; 4 - симазин;
5 - семерон; 6 - мезоранил; 7 - метазин
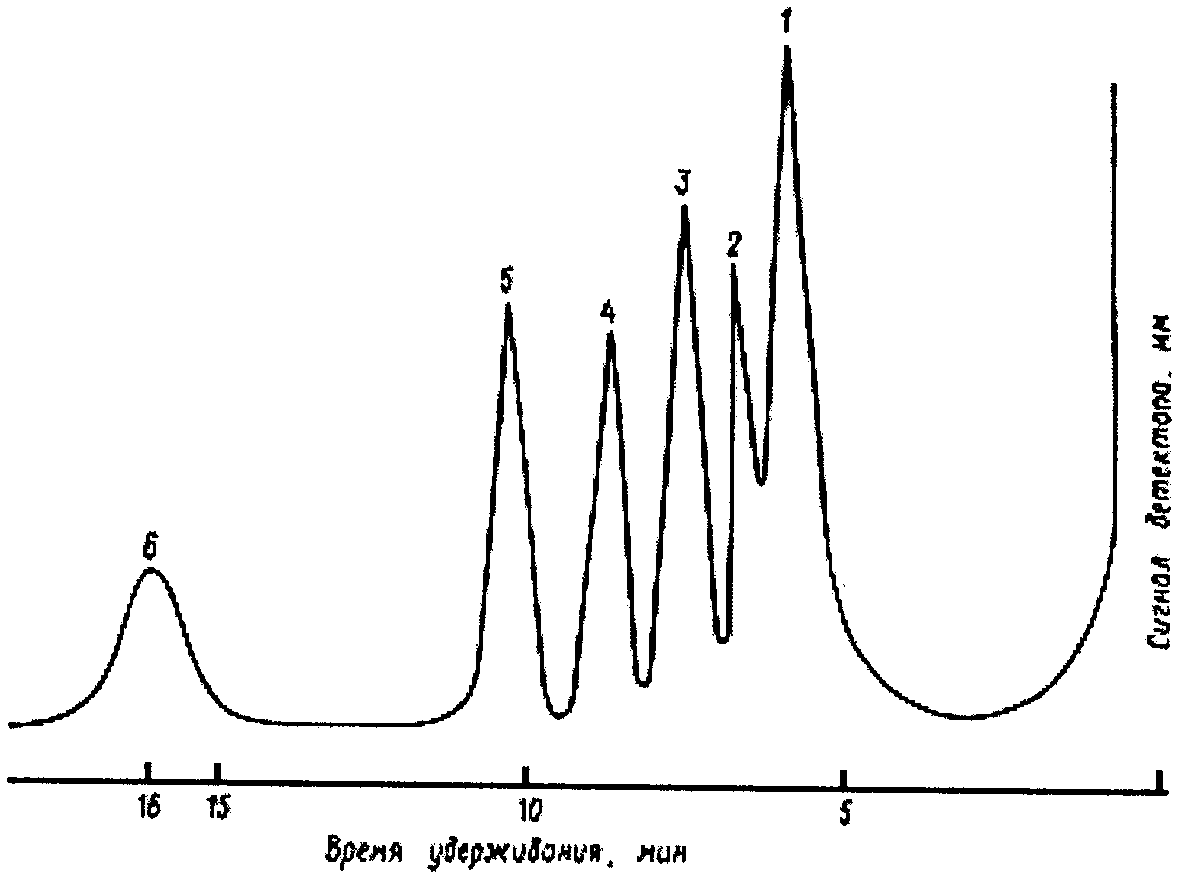
Рис. 25. Хроматограмма смеси симм-триазиновых гербицидов
на колонке длиной 2 м с НЖФ SE-30 (5%)
1 - атразин, пропазин, симазин; 2 - тербуметон;
3 - мезоранил; 4 - семерон; 5 - прометрин; 6 - метазин
Таблица 43
Относительные времена удерживания <*>
симм-триазиновых гербицидов (по пропазину)
при хроматографировании на НЖФ различной полярности
--------------------------------
<*> Время удерживания получено при анализе симм-триазиновых гербицидов на хроматографе "Хром-5" (Чехо-Словакия).
Наименование гербицида | НЖФ | |
карбовакс 20 М (5%) (колонка I <**>) | SE-30 (5%) (колонка II <**>) | |
Тербуметон | 0,83 | 1,16 |
Пропазин | 1,00 | 1,00 |
Атразин | 1,41 | 1,00 |
Прометрин | 1,52 | 1,86 |
Котофор | 1,55 | - |
Симазин | 1,92 | 0,97 |
Семерон | 2,25 | 1,56 |
Мезоранил | 4,08 | 1,33 |
Метазин | 6,22 | 2,91 |
--------------------------------
<**> Параметры колонок приведены в табл. 42.
Времена удерживания некоторых гербицидов на основной колонке I практически совпадают, поэтому в случае их одновременного присутствия в пробе морской воды эти гербициды будут выходить на хроматограмме одним пиком. В случае необходимости их можно разделить на колонке с неполярной жидкой фазой, например SE-30 (5%) (используемой также для анализа хлорорганических пестицидов). Для этого необходимо предварительно определить времена удерживания гербицидов на альтернативной колонке аналогично тому, как это делалось на основной.
Содержание симм-триазиновых гербицидов в анализируемой пробе морской воды находят по формуле
 ,
,где Cx - концентрация вещества в пробе, мкг/л; Cст - концентрация соответствующего гербицида в стандартном растворе, мкг/мл; Sx - площадь пика определяемого вещества на хроматограмме пробы морской воды, равная произведению высоты пика на его ширину при h/2, см2; Sст - площадь пика соответствующего гербицида на хроматограмме стандартного раствора, см2; V1 - объем экстракта после концентрирования, мл; V2 - объем пробы морской воды, взятой для анализа, л.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09 по 25.12.89 (табл. 44), настоящая методика определения симм-триазиновых гербицидов в морской воде допущена к применению в организациях Росгидромета.
Таблица 44
РЕЗУЛЬТАТЫ МЕТРОЛОГИЧЕСКОЙ АТТЕСТАЦИИ МВИ
Наименование гербицида | Диапазон концентрации, мкг/л | Показатель воспроизводимости | Показатель правильности | Показатель погрешности МВИ, суммарная погрешность |
Тербуметон | 0,8 - 5,0 | 80 | 25,0 | 26,0 |
5,0 - 10,0 | 3,9 | 9,7 | 10,8 | |
10,0 - 20,0 | 2,6 | 7,2 | 7,8 | |
Пропазин | 1,2 - 5,0 | 7,4 | 17,0 | 19,0 |
5,0 - 10,0 | 6,7 | 14,0 | 16,0 | |
10,0 - 20,0 | 5,2 | 10,6 | 12,2 | |
Атразин | 0,8 - 4,0 | 9,5 | 18,0 | 20,0 |
4,0 - 10,0 | 5,2 | 12,3 | 14,0 | |
10,0 - 20,0 | 3,2 | 6,8 | 7,6 | |
Симазин | 1,0 - 5,0 | 12,0 | 31,0 | 34,0 |
5,0 - 10,0 | 2,4 | 6,0 | 7,0 | |
10,0 - 20,0 | 2,4 | 6,0 | 7,0 | |
Семерон | 1,2 - 5,0 | 8,4 | 24,0 | 26,0 |
5,0 - 10,0 | 5,5 | 11,5 | 13,2 | |
10,0 - 20,0 | 4,6 | 10,3 | 11,5 | |
Мезоранил | 0,8 - 4,0 | 8,8 | 31,0 | 33,5 |
4,0 - 12,0 | 6,0 | 13,0 | 15,0 | |
12,0 - 40,0 | 4,9 | 11,0 | 12,3 | |
Метазин | 0,8 - 4,0 | 9,6 | 29,5 | 32,0 |
4,0 - 12,0 | 8,7 | 18,0 | 20,7 | |
12,0 - 20,0 | 4,9 | 10,2 | 11,6 | |
20,0 - 40,0 | 3,6 | 7,8 | 8,8 |
Анализ проб воды на содержание симм-триазиновых гербицидов должен выполнять опытный, квалифицированный химик-аналитик, владеющий техникой очистки растворителей, проведения экстракции, вакуумной перегонки, знающий основы газовой хроматографии и прошедший соответствующий инструктаж по технике безопасности.
Для анализа 10 проб гербицидов требуется 39,0 чел.-ч, в том числе:
на взятие проб из батометра - 0,5 чел.-ч;
на приготовление растворов реактивов - 4,0 чел.-ч;
на подготовку посуды - 1,5 чел.-ч;
на подготовку прибора к измерениям - 7,0 чел.-ч;
на проведение пробоподготовки - 15,0 чел.-ч;
на выполнение измерений - 10,0 чел.-ч;
на проведение расчетов - 1,0 чел.-ч.
1. Мельников Н.Н., Новожилов К.В., Белан С.Р., Пылова Т.Н. Справочник по пестицидам. М.: Химия, 1985. 352 с.
2. Определение симм-триазиновых гербицидов в морской воде. Методические указания. РД 52.10.181-89. М.: Госкомгидромет, 1989. 30 с.
3. Balley R., Le Bel J., Lawrence J.F. Chromatography of s-Triazines. J. Chrom., 1978, v. 161, N 1, p. 251 - 257.
4. Lee H.B., Stokker Y.D. Analysis of eleven Triazines in natural waters. J. Assoc. of Anal. Chem., 1986, v. 69, N 4, p. 568 - 572.
В современном сельском хозяйстве для борьбы с сорной растительностью на посевах зерновых, хлопчатника, кукурузы, силосных культур в качестве гербицидов широко применяются производные 2,4-дихлорфеноксиуксусной кислоты (2,4-Д) - натриевая и диметиламмониевая соли, а также бутиловый (2,4-ДБЭ) и октиловый эфиры [2]. Гербициды группы 2,4-Д по совокупности свойств относятся к разряду перспективных, поэтому объем их применения постоянно возрастает.
Основная часть гербицидов группы 2,4-Д, как и большинства пестицидов, попадает в морскую среду с речным и материковым стоком, в связи с чем возможно их накопление в устьевых и прибрежных районах морей, особенно весной в период активного применения гербицидов. Известны также случаи прямого внесения эфиров 2,4-Д в водоемы с целью подавления роста водной растительности [4].
Гербициды группы 2,4-Д слабо персистентны: при попадании в водную среду эфиры гидролизуются до свободной феноксикислоты в течение нескольких суток, а соли - двух-трех недель [5]. Сама 2,4-Д достаточно устойчива и сохраняет активность в почве до полугода [1], а время ее жизни в аэрируемой морской воде составляет около четырех месяцев [4].
Применение гербицидов группы 2,4-Д строго ограничено. Натриевая и диметиламмониевая соли 2,4-Д малотоксичны, их ПДК в водоемах рыбохозяйственного назначения составляют соответственно 20 и 100 мкг/л. Эфиры и 2,4-Д среднетоксичны, ПДК 2,4-ДБЭ составляет 4 мкг/л; для других эфиров и свободной 2,4-Д ПДК не разработаны [2].
Наиболее чувствительным, информативным и доступным методом определения гербицидов группы 2,4-Д в объектах окружающей среды является газожидкостная хроматография с детектором электронного захвата и с использованием слабополярных жидких фаз SE-30 (5%), OV-17 (5%), OV-225 (5%) [1, 3].
Предлагаемая методика позволяет определять сумму растворенных и взвешенных форм гербицидов группы 2,4-Д в морских и распресненных водах указанным методом [3].
Сущность метода анализа заключается в последовательном извлечении из одной пробы воды сначала эфиров, а затем солей 2,4-Д в виде свободной кислоты различными органическими растворителями в соответствующих условиях и последующем дифференцированном анализе полученных экстрактов [3].
Экстракт эфиров концентрируют отгонкой растворителя в вакууме до небольшого объема и анализируют методом газожидкостной хроматографии с детектором электронного захвата, например постоянной скорости рекомбинации.
Экстракт, содержащий 2,4-Д, упаривают досуха в вакууме, остаток этерифицируют бутиловым спиртом в присутствии концентрированной серной кислоты, и полученный бутиловый эфир определяют хроматографически с детектором постоянной скорости рекомбинации [1, 3].
Идентификацию исследуемых веществ проводят по времени удерживания. Количественный расчет их содержания проводят методом соотношения с градуировочным раствором по площадям пиков 2,4-Д на хроматограммах градуировочного раствора и пробы воды.
Показатели погрешности измерений рассчитаны для диапазона концентраций 2,4-ДБЭ и натриевой соли 2,4-Д 5 - 47 мкг/л.
Органические соединения, соэкстрагирующиеся с 2,4-ДБЭ, натриевой и диметиламмониевой солями 2,4-Д из проб морской воды, не мешают их газохроматографическому определению.
Для выполнения анализа применяются:
хроматограф любой марки с детектором электронного захвата или постоянной скорости рекомбинации;
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104;
весы лабораторные 4-го класса точности с наибольшим пределом взвешивания 500 г по ГОСТ 24104;
микрошприцы типа МШ-10М вместимостью 10 мкл по ТУ 25-05-2152;
сушильный шкаф, например типа ШС-3;
ротационный испаритель, например типа ИР-1, ИР-1М;
лабораторный pH-метр-милливольтметр "pH-673" по ТУ 25-05-1181-76;
насос водоструйный стеклянный по ГОСТ 25336 или пластмассовый КМ-1230 по ТУ 64-1-862;
печь электрическая муфельная МП-24 по СТУ 102-753;
плитка электрическая с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
секундомер по ГОСТ 5072;
баня водяная по ТУ 46-22-608;
батометр пластмассовый 7-литровый, например системы ИОАН;
баллон газовый для азота по ГОСТ 949;
штатив лабораторный ШЛ с зажимами и кольцами по ТУ 64-1-707;
шланги резиновые по ГОСТ 5496;
шланги вакуумные по ТУ 38-105881;
термометр ТЛ-5 1-А по ГОСТ 215;
колонки хроматографические стеклянные длиной 2,0 м с внутренним диаметром 3 мм по ТУ 25-05-2815-82;
колбы мерные на 25; 50 и 100 мл по ГОСТ 1770;
колбы круглодонные на 500 мл по ГОСТ 25336;
колбы грушевидные на 25 и 250 мл по ГОСТ 25336;
стаканы химические на 400 мл по ГОСТ 25336;
цилиндры мерные на 50 мл по ГОСТ 1770;
воронки химические диаметром 50 - 80 мм по ГОСТ 25336;
воронки делительные на 100; 500 и 1000 мл по ГОСТ 25336;
пипетки на 0,2; 2,0; 5,0 мл по ГОСТ 20292;
пробирки на 10 мл по ГОСТ 1770;
холодильник прямой по ГОСТ 25336;
аллонж, тип АИО, по ГОСТ 25336;
склянки стеклянные с притертой пробкой на 300 и 5000 мл;
аппарат для встряхивания жидкости в лабораторной посуде, например типа АВУ-1, по ТУ 64-1-1081;
фильтры бумажные, тип ФОМ, по ТУ 6-09-1678;
бумага индикаторная универсальная по ТУ 6-09-181;
капилляры стеклянные;
аппарат для перегонки растворителей;
хроматон N-AW-DMCS или N-AW-HMDS зернения 0,125 - 0,160 мм с нанесенной неподвижной жидкой фазой OV-225, SE-30 или ХЕ-60 в количестве 5%;
азот особой чистоты по ГОСТ 9293 или поверочный нулевой газ (ПНГ);
гексан, ч., перегнанный по ТУ 6-09-3375;
изобутиловый (или н-бутиловый) эфир уксусной кислоты (бутилацетат, изобутилацетат), ч.д.а., перегнанный по ТУ 6-09-701;
спирт бутиловый, ч.д.а., "абсолютированный" по ГОСТ 6006;
натрий сернокислый безводный, ч.д.а., по ГОСТ 4166;
кислота серная конц., х.ч. (плотность 1,84), по ГОСТ 4204;
натрия гидроксид, х.ч., по ГОСТ 4328;
калий двухромовокислый, ч.д.а., по ГОСТ 4220;
вода дистиллированная по ГОСТ 6709;
2,4-дихлорфеноксиуксусной кислоты бутиловый эфир (2,4-ДБЭ) по ТУ 6-09-12-163 или стандартный раствор 2,4-ДБЭ в гексане;
2,4-дихлорфеноксиуксусной кислоты натриевая соль по ТУ 6-09-12-148 или стандартный раствор в гексане.
Пробу морской воды объемом 5 л отбирают с поверхностного горизонта эмалированным ведром или стеклянной бутылью объемом 5 - 10 л, помещенной в проволочную обрешетку, к которой снизу прикреплен груз массой 5 кг. Для отбора проб морской воды с горизонтов на небольших глубинах допускается применение 7-литрового пластмассового батометра, однако во избежание загрязнения пробы воды пластификаторами, содержащимися в пластмассе, процесс отбора проб должен занимать не более 30 мин. По этой же причине не допускается хранение пробы в полиэтиленовой посуде с полиэтиленовыми или резиновыми пробками.
Отобранную пробу морской воды без фильтрации помещают в стеклянную бутыль вместимостью 5 л и закрывают стеклянной пробкой.
Пробы воды хранят не более суток при комнатной температуре.
Экстракты в гексане и изобутилацетате хранят в склянках с притертыми пробками в темноте при температуре 5 - 10 °С. Срок хранения до 3 мес.
4.1.1. Сульфат натрия для осушения экстрактов прокаливают в муфельной печи при температуре 300 - 350 °С в течение 7 - 8 ч. Прокаленный сульфат натрия хранят в герметически закупоренной склянке. Срок хранения не ограничен.
4.1.2. Раствор едкого натра концентрацией 1,6 моль/л готовят растворением 6,4 г щелочи в 100 мл воды.
4.1.3. Хромовая смесь готовится перед употреблением растворением двухромовокислого калия (9,9 г) в концентрированной серной кислоте (100 мл).
4.1.4. Раствор детергентов готовят растворением 10 г любого синтетического моющего средства в 1 л кипящей воды. Используют свежеприготовленный раствор.
4.1.5. Раствор серной кислоты концентрацией 1 моль/л готовят добавлением 5,3 мл концентрированной серной кислоты к 50 мл дистиллированной воды. После охлаждения раствора его объем доводят до 100 мл.
4.1.6. 10%-ный раствор сульфата натрия готовят растворением 10 г безводного сульфата натрия в 90 мл дистиллированной воды.
4.1.7. Бутиловый спирт сушат над безводным сернокислым натрием не менее суток и перегоняют с дефлегматором, отбирая фракцию с температурой кипения 114 - 118 °С. Срок хранения 6 мес. при температуре 5 - 10 °С.
4.1.8. Гексан перед употреблением перегоняют, собирая фракцию с температурой кипения 68 - 70 °С. Срок хранения 1 год.
4.1.9. Изобутиловый (или н-бутиловый) эфир уксусной кислоты подготавливают следующим образом: 1 л реактива помещают в делительную воронку, добавляют 15 мл раствора едкого натра концентрацией 1,6 моль/л и встряхивают в течение 3 мин. Водный слой отбрасывают, эфир промывают дистиллированной водой до нейтральной реакции промывных вод, сушат над безводным сульфатом натрия и перегоняют изобутилацетат, отбирая фракцию с температурой кипения 116 - 117 °С (и с температурой кипения 124 - 125 °С для н-бутилацетата). Срок хранения 1 год.
Стеклянная посуда промывается в следующем порядке: горячим раствором детергентов, дистиллированной водой (трижды), хромовой смесью, дистиллированной водой (5 - 7 раз), ацетоном. После промывания посуда сушится при 150 - 200 °С в течение 2 - 3 ч.
4.3.1. 2,4-ДБЭ
Пробу воды объемом 3 - 5 л помещают в стеклянную бутыль вместимостью 5 л и экстрагируют 2,4-ДБЭ гексаном в нейтральной или слабощелочной среде трижды по 5 мин вручную или с помощью аппарата для встряхивания жидкости в лабораторной посуде. При каждой экстракции расходуется 20 мл гексана на литр пробы морской воды.
После окончания экстракции в объединенные экстракты добавляют безводный сульфат натрия для осушения и выдерживают при комнатной температуре в вытяжном шкафу не менее суток.
4.3.2. Натриевая соль 2,4-Д
К пробе морской воды, оставшейся после экстракции из нее 2,4-ДБЭ, добавляют по каплям раствор серной кислоты концентрацией 1 моль/л до установления pH = 2. Образующуюся феноксикислоту (2,4-Д) экстрагируют трижды по 5 мин изобутилацетатом (или н-бутилацетатом) порциями по 20 мл растворителя на литр пробы.
После окончания экстракции объединенные экстракты высушивают над безводным сульфатом натрия не менее суток.
Высушенный гексановый экстракт фильтруют небольшими порциями в круглодонную колбу вместимостью 500 мл и отгоняют растворитель в вакууме водоструйного насоса в токе азота или гелия на водяной бане при температуре 35 - 37 °С до объема 2 - 3 мл. Применение вакуумной смазки или других смазочных материалов не допускается. Сконцентрированный экстракт количественно переносят стеклянным капилляром в пробирку на 5 мл и измеряют его объем. Полученный раствор используют для анализа на хроматографе. Допускается хранение экстракта в течение месяца при температуре 0 - 4 °С.
этерификации 2,4-Д
Высушенный экстракт 2,4-Д в изобутилацетате (н-бутилацетате) фильтруют в круглодонную колбу на 500 мл и отгоняют растворитель в вакууме водоструйного насоса в токе азота (или инертного газа) на водяной бане температурой 55 - 60 °С до объема 10 - 15 мл. Остаток количественно переносят в грушевидную колбу на 25 мл и отгоняют изобутилацетат досуха в аналогичных изложенным выше условиях.
К сухому остатку, полученному в колбе после отгонки изобутилацетата, добавляют сначала 1 мл бутилового спирта, затем 0,5 мл концентрированной серной кислоты. Колбу с реакционной смесью закрывают притертой стеклянной пробкой и помещают на 20 мин на кипящую водяную баню. После этого колбу охлаждают до комнатной температуры и экстрагируют полученный 2,4-ДБЭ гексаном (4 мл) дважды по 2 мин. К объединенному гексановому экстракту добавляют 5 мл 10%-ного раствора сульфата натрия, энергично встряхивают 1 - 2 мин, затем водный слой отбрасывают, а гексановый экстракт фильтруют через 5 - 7 г безводного сульфата натрия в грушевидную колбу на 25 мл. Делительную воронку ополаскивают порцией гексана 2 - 3 мл, промывают этим же раствором сульфат натрия. Растворитель отгоняют в вакууме водоструйного насоса <*> в токе азота (или инертного газа) на водяной бане с температурой 35 - 37 °С до конечного объема 2 - 3 мл, количественно переносят с помощью стеклянного капилляра в мерную пробирку на 5 мл и используют для анализа на хроматографе. Допускается хранение экстракта в течение месяца при температуре 0 - 4 °С.
--------------------------------
<*> Для отгонки растворителя возможно использование ротационного испарителя.
4 - 6 мкл экстракта, полученного согласно п. п. 4.4, 4.5, вводят в испаритель хроматографа и записывают хроматограмму. После выхода пика 2,4-ДБЭ прибор оставляют в холостом режиме работы на 40 - 50 мин во избежание возможного оседания на колонке органических высокомолекулярных примесей, содержащихся в морской воде.
Холостое определение проводят перед анализом проб воды. Цель определения - проверить чистоту реактивов и материалов, используемых для анализа. Для выполнения холостого определения берут тот же объем экстрагента (гексана, изобутилацетата), раствора серной кислоты, концентрацией 1 моль/л бутилового спирта и 10%-ного раствора сульфата натрия, что и для анализа одной пробы воды, и проводят с ними последовательно все операции, как описано в п. п. 4.3, 4.4, 4.5.1 и 4.5.2.
При отсутствии пиков на хроматограмме холостого опыта холостое определение повторяют для каждой новой партии реактивов.
Если же на хроматограмме холостого опыта появляются пики с временами удерживания, близкими к временам удерживания исследуемых веществ, то необходимо путем постадийного исследования установить, какой из реактивов загрязнен, и заменить его таким же реактивом из другой партии.
Для проверки чистоты используемой посуды ее ополаскивают 3 мл гексана и 4 - 6 мкл полученного смывного раствора вводят в испаритель хроматографа. Отсутствие на хроматограмме пиков (кроме пика, соответствующего растворителю) служит доказательством чистоты посуды.
6.1.1. Для приготовления стандартного раствора 2,4-ДБЭ ампулу со стандартным образцом (3 мг в 1 мл гексана) вскрывают и содержимое быстро переносят в мерную колбу вместимостью 50 мл с притертой пробкой. Ампулу промывают 2 - 3 раза гексаном и сливают в мерную колбу со стандартным образцом, после чего доводят гексаном ее до метки. Полученный раствор имеет концентрацию 60 мкг/мл.
Раствор стабилен при хранении в холодильнике в течение 6 месяцев.
6.1.2. Рабочий стандартный раствор 2,4-ДБЭ готовят непосредственно перед работой. Для этого 1 мл стандартного раствора 2,4-ДБЭ помещают в мерную колбу вместимостью 50 мл с притертой пробкой и доводят гексаном до метки. Полученный раствор имеет концентрацию 1,2 мкг/мл.
В хроматограф при условиях, подобранных согласно п. 6.4, вводят 6 мкл стандартного рабочего раствора 2,4-ДБЭ и определяют его время удерживания. Затем стандартный раствор разбавляют в 2; 4 и 8 раз. Для этого в мерные колбы вместимостью 25; 25 и 50 мл вносят соответственно 12,5; 6,25 и 6,25 мл рабочего стандартного раствора и доводят содержимое колбы до метки гексаном. Полученные растворы имеют концентрацию 0,6; 0,3 и 0,15 мкг/л. По 6 мкл каждого раствора вводят в испаритель хроматографа и записывают хроматограммы. Проводят количественный обсчет всех полученных хроматограмм и, построив для каждого компонента график зависимости площади пика от количества вещества, убеждаются в ее линейности для данного диапазона концентраций.
Стеклянную колонку длиной 1,8 - 2,0 м и диаметром 2,5 мм промывают этиловым спиртом и ацетоном, сушат и заполняют готовым носителем с нанесенной жидкой фазой. Для этого один конец колонки плотно закрывают небольшим тампоном из стекловаты, промытой ацетоном, затем кусочком марли и подсоединяют к водоструйному насосу. После этого через свободный конец заполняют колонку насадкой, прибавляя ее небольшими порциями и уплотняя постукиванием по колонке палочкой с резиновым наконечником при постоянно работающем насосе. Необходимо следить за тем, чтобы насадка ложилась равномерно, без пустот, с одинаковой плотностью. Заполненную колонку закрывают тампоном из стекловаты и помещают в термостат колонок хроматографа, не подсоединяя к детектору. Кондиционируют колонку в токе газа-носителя при расходе его 15 - 20 мл/мин при температуре 100 °С в течение 6 ч и при температуре 200 °С в течение 6 - 8 ч.
Подготовку хроматографа проводят в соответствии с инструкцией по эксплуатации.
Устанавливают с помощью пенного расходомера расход газа-носителя (азот) 25 - 28 мл/мин. Подсоединяют колонку к детектору и проверяют герметичность соединений, нанося на них кисточкой мыльную пену.
Устанавливают поступление в детектор азота со скоростью 115 - 125 мл/мин. Измеряют суммарный расход азота на выходе прибора и контролируют ежедневно перед началом работы.
Устанавливают температуру термостата колонок 175 - 190 °С, испарителя 220 - 230 °С, термостата детектора 240 - 250 °С, устанавливают рабочий диапазон измерений на шкале электрометра и скорость диаграммной ленты (0,4 - 0,6 см/мин). При работе в рабочем диапазоне измерений электрометра высота пика 2,4-ДБЭ, соответствующая 4 мкл стандартного раствора с концентрацией 1,2 мкг/мл, должна составить не менее 1/3 шкалы самописца.
Критерием полноты кондиционирования газохроматографической колонки является соответствие дрейфа и нерегулярных шумов нулевой линии паспортным данным прибора.
Для насыщения колонки исследуемыми соединениями вкалывают подряд 5 - 7 раз по 6 мкл стандартного раствора 2,4-ДБЭ с концентрацией 1,2 мкг/мл, после чего колонка полностью готова для анализа.
Определение параметров колонки и детектора проводят аналогично описанному в гл. "Симм-триазиновые гербициды".
Для определения характеристик линейности диапазона детектирования готовят стандартные растворы 2,4-ДБЭ шести концентраций, различающихся не более чем в два раза. По 4 - 6 мкл полученных растворов инжектируют в испаритель хроматографа и записывают хроматограммы на рабочей шкале электрометра. Рассчитывают площади пиков на полученных хроматограммах и определяют отношение концентрации 2,4-ДБЭ к площади пиков (K = C/S). Линейность детектирования сохраняется для концентраций, при которых значения K отличаются не более чем на 5%: [(K2 - K1)/K2]·100% <= 5%. В случае отсутствия линейности детектирования строят градуировочный график для всех используемых диапазонов чувствительности.
В испаритель хроматографа вводят микрошприцем 4 - 6 мкл рабочего стандартного раствора 2,4-ДБЭ и записывают хроматограмму. Время удерживания рассчитывают по трем результатам хроматографирования. Этот параметр необходимо проверять ежедневно перед началом определений после выхода прибора на режим.
Затем вводят в испаритель 4 - 6 мкл экстракта проб, подготовленных согласно п. п. 4.3, 4.4 и 4.5, 2,4-ДБЭ идентифицируют, сравнивая времена удерживания компонентов пробы морской воды с соответствующим параметром пика на хроматограмме стандартного раствора.
Условия хроматографирования смеси эфиров 2,4-Д приведены в табл. 45.
Таблица 45
Условия <*> хроматографирования смеси эфиров 2,4-Д
--------------------------------
<*> Условия приведены для газового хроматографа "Цвет-110".
Параметр | Значение |
1. Длина хроматографической колонки, м | 2,0 |
2. Жидкая фаза, нанесенная на твердый носитель | OV-225 (5%) на хроматоне N-супер зернением 0,16 - 0,20 мм |
3. Расход газа-носителя (азота), см3/мин | |
через колонку | 25 - 28 |
через детектор | 115 - 125 |
4. Температурный режим, °С | |
колонки | 165 - 175 |
испарителя | 225 - 235 |
детектора | 245 - 255 |
5. Рабочая шкала электрометра, А | 10 х 10-12 |
6. Скорость протяжки диаграммной ленты, мм/ч | 240 |
Типичная хроматограмма некоторых эфиров 2,4-Д представлена на рис. 26. Времена удерживания относительно 2,4-ДБЭ приведены в табл. 46.

Рис. 26. Хроматограмма смеси эфиров 2,4-Д
на колонке длиной 1,8 м с НЖФ OV-225 (5%)
1 - метиловый эфир; 2 - этиловый эфир; 3 - бутиловый эфир;
4 - пик системы; 5 - октиловый эфир
Таблица 46
Времена удерживания (относительно 2,4-ДБЭ) эфиров 2,4-Д
Эфир 2,4-Д | Относительное время удерживания |
Метиловый | 0,44 |
Этиловый | 0,52 |
Бутиловый | 1,00 |
Октиловый | 1,76 |
8.1.1. 2,4-ДБЭ
Содержание 2,4-ДБЭ в анализируемой пробе морской воды находят по формуле

где Cx - концентрация 2,4-ДБЭ в пробе, мкг/л; Cст - концентрация 2,4-ДБЭ в стандартном растворе, мкг/мл; Sx - площадь пика 2,4-ДБЭ на хроматограмме пробы морской воды, равная произведению высоты пика h на его ширину на уровне h/2, см2; V1 - объем экстракта после концентрирования, мл; Vст - объем стандартного раствора, вводимого в испаритель хроматографа, мкл; Sст - площадь пика 2,4-ДБЭ на хроматограмме стандартного раствора, см2; V2 - объем пробы морской воды, взятой для анализа, л; Vx - объем экстракта пробы, вводимого в испаритель хроматографа, мкл.
8.1.2. Натриевая соль 2,4-Д
Содержание натриевой соли 2,4-Д в анализируемой пробе морской воды находят по формуле
 ,
,где Cy - концентрация натриевой соли 2,4-Д в пробе морской воды, мкг/л; Cx - концентрация 2,4-ДБЭ, полученного этерификацией натриевой соли 2,4-Д согласно п. 4.5.2, рассчитанная по формуле (1), мкг/л; M2 - молекулярная масса 2,4-ДБЭ; M1 - молекулярная масса натриевой соли 2,4-Д; Kэ - коэффициент этерификации натриевой соли 2,4-Д в условиях, описанных в п. 4.5.2, равный 0,76.
Таким образом,
Cy = 1,2·Cx.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09 по 20.12.1989 (табл. 47), настоящая методика определения гербицидов группы 2,4-Д в морской воде допущена к применению в организациях Росгидромета.
Таблица 47
Результаты метрологической аттестации МВИ
Вещество | Диапазон концентрации, мкг/л | Показатель вос- производимости | Показатель правильности | Показатель погрешности МВИ |
2,4-ДБЭ | 9,5 - 19,0 | 2,0 | 5,2 | 5,8 |
19,1 - 47,5 | 1,8 | 3,7 | 4,2 | |
Натриевая соль | 5,0 - 20,0 | 3,0 | 7,3 | 8,2 |
2,4-Д | 20,1 - 30,0 | 2,0 | 7,4 | 7,8 |
Анализ проб воды на содержание гербицидов группы 2,4-Д должен выполняться опытным, квалифицированным химиком-аналитиком, владеющим техникой очистки растворителей, проведения экстракции, вакуумной перегонки, знающим теоретические основы и технику газовой хроматографии и прошедшим соответствующий инструктаж по технике безопасности.
Для анализа 10 проб гербицидов группы 2,4-Д в морской воде требуется 82,0 чел.-ч, в том числе:
на взятие проб из батометра - 0,5 чел.-ч;
на приготовление растворов реактивов и очистку растворителей - 20 чел.-ч;
на подготовку посуды - 4 чел.-ч;
на подготовку прибора к измерениям - 4 чел.-ч;
на проведение пробоподготовки - 44 чел.-ч;
на выполнение измерений - 8 чел.-ч;
на проведение расчетов - 1,5 чел.-ч.
1. Бабкина Э.И., Алексеева Л.Б., Трублаевич Ж.Н. Газохроматографическое определение 2,4-дихлорфеноксиуксусной кислоты в почве в виде этилового эфира. ЖАХ. 1985, т. 40, N 2, с. 2048 - 2051.
2. Мельников Н.Н., Новожилов К.Б., Белан С.Р., Пылова Г.В. Справочник по пестицидам. М.: Химия, 1985. 350 с.
3. Определение гербицидов группы 2,4-Д в морской воде. Методические указания. РД 52.10.239-90. М.: изд. Госкомгидромета, 1990. 31 с.
4. Birmingham B.C., Colman B. Persistence and fate of 2,4-D butoxyethanol ester in artificial ponds. J. Environ. Qual., 1985, v. 14, N 1, p. 100 - 104.
5. Alexander M.C., Gerrich F.M., Mayes M.A. Acute toxicity of four phenoxy herbicides to aquatic organisms. Bull. Environ. Contam. Toxicol., 1986, v. 35, N 3, p. 314 - 321.
Ксантогенаты относятся к числу флотореагентов, применяющихся в качестве коллекторов при флотации руд цветных металлов. Ксантогенаты являются токсичными веществами и их предельно допустимая концентрация для вод водоемов составляет 1 мкг/л.
Экстракционно-фотометрические [1, 2] и титриметрические [4] методики позволяют определять относительно высокие, более 50 мкг/л, концентрации этих веществ в пробах. С помощью настоящей методики можно находить общее содержание ксантогенатов из пробы воды объемом 250 мл в диапазоне концентраций от 1 до 30 мкг/л [3].
Ксантогенаты не устойчивы в водных растворах и довольно быстро гидролизуются, поэтому предлагаемую методику рекомендуется применять для анализа вод морских водоемов, расположенных в непосредственной близости от соответствующих горнообогатительных комбинатов.
Определение ксантогенатов основано на образовании ксантогената меди (I), экстракции его из пробы воды хлороформом и реэкстракции меди (I) в азотную или соляную кислоту. По атомной абсорбции меди в аликвоте реэкстракта судят о содержании ксантогенатов в пробе. Минимально определяемая концентрация составляет 1 мкг/л. Мешать определению могут дитиофосфаты, также образующие экстрагируемые соединения с медью (I). В отличие от легко гидролизующихся ксантогенатов дитиофосфаты устойчивы в водных средах, поэтому разрушить их в растворе, не затрагивая при этом ксантогенатов, не представляется возможным. Наиболее целесообразным в данном случае является учет влияния дитиофосфатов. Для этого пробу делят на две части, в первой определяют суммарное содержание ксантогенатов и дитиофосфатов, во второй, после подкисления и полного гидролиза ксантогенатов, - только дитиофосфаты. Разница в полученных аналитических сигналах соответствует содержанию ксантогенатов в пробе. Если заведомо известно, что проба не содержит дитиофосфатов, то анализ проводится только по схеме, предусмотренной для первой части.
Для выполнения анализа применяются:
атомно-абсорбционный спектрофотометр с непламенной атомизацией проб любой марки;
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104;
центрифуга с набором центрифужных пробирок, например типа ЦЕЛ-1, по ТУ 5-375-4166;
электроплитка с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
баллон газовый для азота (гелия) по ГОСТ 949;
колбы мерные 2-го класса точности исполнения 2 вместимостью 100; 250; 500 мл по ГОСТ 1770;
воронки делительные на 500 мл по ГОСТ 25336;
пипетки на 2; 5; 10 и 20 мл по ГОСТ 20292;
стаканы на 400 мл по ГОСТ 25336;
цилиндры мерные на 50 мл по ГОСТ 1770;
колбы лабораторные конические со шлифом на 25 мл по ГОСТ 25336;
воронки химические типа В диаметром 50 - 80 мм по ГОСТ 25336;
бюретки с двухходовым краном и автоматическим нулем на 25 мл по ГОСТ 20292;
колбы короткогорлые со шлифом на 250 мл по ГОСТ 25336;
бюксы по ГОСТ 25336;
фильтры бумажные, тип ФОМ, по ТУ 6-09-1678;
вода дистиллированная по ГОСТ 6709;
ксантогенат калия этиловый, ч.д.а., по ТУ 609-830 или технический по ГОСТ 7927;
медь хлористая, ч., по ГОСТ 4164;
кислота уксусная, ч., по ГОСТ 61;
кислота соляная, ч.д.а., по ГОСТ 3118;
кислота азотная, ч.д.а., по ГОСТ 4461;
натрий уксуснокислый трехводный, ч.д.а., по ГОСТ 199;
хлороформ, х.ч., по ТУ 6-09-06-800;
калия гидроксид, ос.ч., по ОСТ 6-01-301;
барий хлористый, ч.д.а., по ГОСТ 4108;
иод, ч.д.а., по ГОСТ 4159;
крахмал растворимый по ГОСТ 10163;
кальций углекислый, ч.д.а., по ГОСТ 4530;
натрий серноватистокислый 5-водный, ч.д.а., по СТ СЭВ 223;
фенолфталеин, ч., по ГОСТ 5850;
калий иодистый, х.ч., по ГОСТ 4232;
эфир диэтиловый медицинский;
ацетон, ос.ч., по ТУ 6-09-3513;
бензол, х.ч., по ТУ 6-09-779;
спирт этиловый ректификат высший сорт по ГОСТ 18300;
азот особой чистоты по ГОСТ 9293 или поверочный нулевой газ (ПНГ).
Пробу морской воды объемом 500 мл отбирают чистым пластмассовым или стеклянным батометром, после чего сразу приступают к ее обработке. Во избежание гидролиза ксантогенатов пробу хранить не рекомендуется. Если окончание анализа (измерение атомной абсорбции) нельзя провести сразу, следует довести первичную обработку пробы до стадии реэкстракции включительно. Устойчивость реэкстрактов сохраняется в течение длительного времени при условии хранения в чистых склянках с плотными пробками.
4.1.1. Раствор уксусной кислоты концентрацией 1 моль/л готовят растворением 30 мл ледяной уксусной кислоты в дистиллированной воде и доведением объема до 500 мл. Раствор устойчив.
4.1.2. Для приготовления раствора ацетата натрия концентрацией 1 моль/л растворяют 136,1 г ацетата натрия в дистиллированной воде и доводят объем до 1 л. Раствор устойчив.
4.1.3. Ацетатный буферный раствор готовят, смешивая 158 мл раствора ацетата натрия и 42 мл раствора уксусной кислоты и разбавляя смесь дистиллированной водой до объема 1 л. Раствор устойчив.
4.1.4. Для приготовления раствора хлористой меди концентрацией 0,25 моль/л растворяют 2,55 г хлористой меди в 10 мл концентрированной соляной кислоты и доводят объем раствора дистиллированной водой до 100 мл. Раствор готовят непосредственно перед обработкой проб. Устойчив в течение 6 - 8 ч при хранении в закрытой склянке <*>.
--------------------------------
<*> Кристаллы хлористой меди белого цвета, однако при стоянии на воздухе быстро зеленеют из-за образования основной соли, поэтому для анализа используют реактив зеленого цвета, который дополнительной очистке не подлежит.
4.1.5. Для приготовления раствора бария хлористого 10%-ного растворяют 10 г хлористого бария в дистиллированной воде и доводят объем до 100 мл.
4.1.6. Для приготовления раствора иода концентрацией 0,25 моль/л сначала растворяют 20 - 22 г иодистого калия в 30 мл дистиллированной воды, затем в полученный раствор добавляют 3,17 г иода. После полного растворения иода доводят объем раствора дистиллированной водой до 500 мл. Раствор хранится в темной склянке с хорошо притертой пробкой.
4.1.7. Раствор соляной кислоты концентрацией 0,1 моль/л готовят растворением 8,3 мл концентрированной соляной кислоты в дистиллированной воде с доведением объема до 100 мл. Можно также готовить раствор соляной кислоты, пользуясь фиксаналами.
4.1.8. Раствор соляной кислоты концентрацией 0,05 моль/л готовят разбавлением 50 мл соляной кислоты концентрацией 0,1 моль/л дистиллированной водой до 100 мл.
4.1.9. Для приготовления раствора крахмала 0,5%-ного встряхивают 0,5 г крахмала в 15 - 20 мл дистиллированной воды. Полученную смесь постепенно вливают в 80 мл кипящей воды и кипятят несколько минут до просветления раствора. Раствор консервируют добавлением 1 капли хлороформа.
4.1.10. Раствор натрия серноватистокислого концентрацией 0,05 моль/л готовят растворением 7,2 г соли в дистиллированной воде, предварительно прокипяченной и охлажденной до комнатной температуры. Доводят объем раствора до 500 мл. Раствор консервируют добавлением 0,5 мл хлороформа и хранят в темной склянке, снабженной поглотительной трубкой с гранулированной щелочью. Лучше готовить раствор из фиксанала.
4.1.11. Раствор фенолфталеина 0,1%-ный готовят растворением 0,1 г фенолфталеина в 100 мл этилового спирта.
4.1.12. Раствор гидроксида калия концентрацией 10 моль/л готовят растворением 56 г щелочи в дистиллированной воде с доведением объема до 100 мл.
При хранении ксантогенаты постепенно подвергаются гидролизу, особенно во влажных помещениях, поэтому необходимо проводить их очистку перед построением градуировочного графика. Для этого ксантогенат сначала перекристаллизовывают из этилового спирта, затем растворяют в горячем ацетоне и осаждают из раствора бензолом. Кристаллы фильтруют через бумажный фильтр, промывают диэтиловым эфиром и сушат на воздухе. Хорошо высушенную соль хранят в бюксе с притертой крышкой.
ксантогенате калия (ГОСТ 7927)
4.3.1. Приготовление раствора ксантогената
Около 1 г очищенного ксантогената взвешивают с погрешностью не более 0,001 г, помещают в мерную колбу на 100 мл и растворяют в дистиллированной воде. Добавляют 10 мл раствора хлористого бария, после чего доводят объем раствора до метки. Дают отстояться, затем фильтруют раствор через бумажный фильтр в сухую колбу. Первую порцию фильтрата (около 20 мл) отбрасывают, остальной фильтрат немедленно используют для анализа.
4.3.2. Определение содержания свободного едкого кали
Отбирают пипеткой 25 мл раствора ксантогената, помещают в коническую колбу на 250 мл, прибавляют 2 - 3 капли раствора фенолфталеина и титруют раствором соляной кислоты концентрацией 0,05 моль/л до исчезновения розового окрашивания. Хорошо очищенный ксантогенат обычно содержит ничтожно малое количество свободной щелочи, поэтому розовой окраски индикатора может не появиться.
4.3.3. Проведение анализа
Раствор, нейтрализованный соляной кислотой, быстро титруют раствором иода в присутствии крахмала. Затем пипеткой отбирают 25 мл (новую аликвоту ксантогената калия), помещают в коническую колбу вместимостью 250 - 300 мл, добавляют к этиловому ксантогенату калия 50 мл (а к бутиловому 25 мл) раствора соляной кислоты концентрацией 0,1 моль/л, закрывают пробкой и выдерживают 15 мин (бутиловый - 30 мин). После этого быстро добавляют около 0,3 г углекислого кальция (на дне колбы должно оставаться его небольшое количество), затем приливают 10 мл раствора иода и титруют раствором тиосульфата натрия до тех пор, пока раствор не примет желтого окрашивания, прибавляют несколько капель раствора крахмала и дотитровывают до исчезновения окрашивания.
4.3.4. Обработка результатов
Содержание основного вещества (C) вычисляют по формуле
 ,
,где V1 - объем раствора иода концентрацией 0,025 моль/л, израсходованный на титрование, мл; V2 - объем раствора тиосульфата натрия концентрацией 0,05 моль/л, V3 - объем раствора иода концентрацией 0,025 моль/л, взятый для анализа и равный 10 мл; m - масса навески продукта, г; K - количество ксантогената калия, соответствующее 1 мл раствора иода концентрацией 0,025 моль/л (для этилового ксантогената K = 0,008015; для бутилового - 0,00942).
Допустимые расхождения между результатами двух параллельных определений не должны превышать 0,5%.
Обычно в очищенном препарате содержится не менее 95% основного вещества. Для получения более высокого содержания очистку можно повторить.
Методика определения ксантогенатов требует большой тщательности выполнения. Особое внимание следует уделить склянкам для сбора экстрактов. Необходимо проводить строгий контроль их чистоты. Для этого предварительно вымытую склянку (коническую колбочку со шлифом) ополаскивают 3 - 5 мл разбавленной азотной или соляной кислотой, после чего промывную кислоту анализируют на атомно-абсорбционном спектрофотометре. Если сигнал абсорбции превышает значение 0,010, то склянку замачивают в разбавленной кислоте на несколько часов, затем ополаскивают несколько раз и снова проверяют на чистоту. При необходимости процедуру очистки повторяют до тех пор, пока значение абсорбции не станет меньше указанного.
Пробу объемом 500 мл делят пополам. Первую ее часть объемом 250 мл переносят в делительную воронку на 500 мл, добавляют 3 мл раствора хлористой меди и 20 мл ацетатного буфера. После перемешивания раствора добавляют в воронку 10 мл хлороформа и экстрагируют в течение 3 мин. После разделения слоев (через 7 - 10 мин) нижний органический слой собирают в склянку (колбочку) емкостью 25 мл, фильтруя его через кусочек ваты, предварительно вымытый хлороформом и высушенный. Если после экстракции образовалась устойчивая эмульсия, то ее собирают в центрифужную пробирку и центрифугируют 10 мин при скорости 3000 об./мин. Содержимое пробирки вновь возвращают в делительную воронку и собирают отделившийся экстракт, как указано выше. Необходимо строго следить, чтобы ни одна капля водной фазы не попала в склянку. Склянки должны быть чистыми и сухими. Затем добавляют к экстракту 200 мкл концентрированной азотной или соляной кислоты, закрывают пробкой и встряхивают несколько раз, после чего добавляют еще 2 мл дистиллированной воды и снова встряхивают в течение 1 мин. После разделения слоев верхний водный слой (реэкстракт) подготовлен к измерению.
Вторую часть пробы также переносят в делительную воронку, добавляют 3 мл раствора хлористой меди и для разрушения ксантогенатов добавляют 1 мл концентрированной соляной кислоты. После 20-минутной выдержки раствор нейтрализуют 1 мл раствора гидроксида калия, добавляют 20 мл ацетатного буфера (pH должен быть таким же, как и в первой части пробы). Последующую обработку проводят, как указано выше.
Для выполнения холостого определения 250 мл дистиллированной воды проводят через все стадии анализа, предусмотренные для первой части пробы. При соблюдении всех требований, предъявляемых к чистоте посуды и реактивов, значения атомной абсорбции холостых определений не превышают обычно 0,010 - 0,015. Холостое определение проводят перед построением градуировочного графика и повторяют для каждой новой партии реактивов.
Основной стандартный раствор этилксантогената калия готовят, растворяя навеску около 0,5 г (с учетом найденного содержания основного вещества) в дистиллированной воде, затем добавляют пять капель раствора гидроксида калия и доводят объем до 100 мл дистиллированной водой. Концентрация этилксантогената калия в основном стандартном растворе 5 г/л. Раствор хранят в холодильнике не более двух недель.
Расчет необходимой навески производят по формуле
 ,
,где C - найденное содержание основного вещества, %.
Например, если содержание основного вещества в этилксантогенате калия составляет 95%, то масса навески - 0,5263 г.
Для приготовления рабочего стандартного раствора этилксантогената калия 0,25 мл основного раствора помещают в мерную колбу на 500 мл и доводят объем до метки дистиллированной водой. Концентрация этилксантогената калия в рабочем стандартном растворе 2,5 мг/л. Раствор готовят в день построения градуировочного графика.
Для приготовления градуировочных растворов в ряд мерных колб объемом 250 мл вносят 0,1; 0,5; 1,0; 1,5; 2,0; 3,0 мл рабочего стандартного раствора ксантогената и доводят объемы растворов до метки дистиллированной водой. Полученные концентрации составляют соответственно 1; 5; 10; 15; 20; 30 мкг/л. Обработку каждого градуировочного раствора производят так же, как обработку первой части пробы при проведении анализа. Каждый градуировочный раствор готовят параллельно не менее трех раз.
Градуировочный график строят по средним значениям измеренных величин атомной абсорбции в координатах "атомная абсорбция - концентрация ксантогенатов в пробе", при этом из каждого значения атомной абсорбции градуировочного раствора вычитают значение атомной абсорбции холостой пробы.
Рабочие параметры и температурно-временной режим атомизации проб устанавливают в соответствии с инструкцией прибора.
После разделения слоев отбирают из верхнего водного слоя 20 мкл с помощью пипетки Эппендорфа и вводят в камеру атомизации атомно-абсорбционного спектрофотометра. Интенсивность абсорбции меди измеряют при 324,7 нм, используя следующий температурно-временной режим работы:
сушка при 80 °С - 30 с;
обжиг при 900 °С - 20 с;
атомизация при 2500 °С - 10 с.
Для каждой пробы получают два значения атомной абсорбции. Первое, полученное в результате обработки первой части пробы (A1), соответствует суммарному содержанию ксантогенатов и дитиофосфатов. Второе, полученное после обработки второй части пробы (A2), соответствует содержанию только дитиофосфатов. Рассчитывают значение 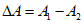 , которое соответствует содержанию ксантогенатов в пробе. По
, которое соответствует содержанию ксантогенатов в пробе. По  с помощью градуировочного графика находят концентрацию ксантогенатов в пробе. Для проб с заведомым отсутствием дитиофосфатов содержание ксантогенатов в пробе находят непосредственно по значению A1, при этом из каждого значения атомной абсорбции необходимо вычесть значение холостого определения.
с помощью градуировочного графика находят концентрацию ксантогенатов в пробе. Для проб с заведомым отсутствием дитиофосфатов содержание ксантогенатов в пробе находят непосредственно по значению A1, при этом из каждого значения атомной абсорбции необходимо вычесть значение холостого определения.
, которое соответствует содержанию ксантогенатов в пробе. По На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09.88 по 25.12.88 (табл. 48), настоящая методика определения ксантогенатов допущена к применению в организациях Росгидромета.
Таблица 48
Результаты метрологической аттестации
Диапазон концентрации ксантогенатов в морской воде, мкг/л | Показатель воспроизводимости | Показатель правильности | Показатель погрешности МВИ, суммарная погрешность |
1 - 10 | 12,3 | 40,0 | 42,0 |
10 - 30 | 2,5 | 7,5 | 8,2 |
К выполнению анализа допускаются лица с высшим или средним специальным образованием, имеющие опыт работы с химическими препаратами и на атомно-абсорбционном спектрофотометре, допущенные к работе с газовыми баллонами.
Для анализа 10 проб требуется 16,8 чел.-ч, в том числе:
на взятие проб из батометра - 0,3 чел.-ч;
на приготовление растворов реактивов - 1 чел.-ч;
на подготовку посуды - 3 чел.-ч;
на подготовку атомно-абсорбционного спектрофотометра к работе - 1,5 чел.-ч;
на проведение экстракции - 6 чел.-ч;
на выполнение измерений - 5 чел.-ч.
1. Кириллова З.П., Мерисов Ю.И. Способ определения ксантогенатов в водных растворах. А. с. 1113721 СССР, Бюллетень изобретений 1984, N 34, МКИ G 21/78.
3. Определение ксантогенатов в морской воде. Методические указания. РД 52.10.182-89. М.: Госкомгидромет, 1989. 19 с.
4. Talegaonkar T., Boparai K.S. Determination of xanthates by reaction with 2,4-dinitrobenzenesulphenyl chloride. J. Indian. Chem. Soc., 1982, v. 59, N 1, p. 109.
Дитиофосфаты являются наиболее характерными собирателями сульфидных минералов при обогащении руд цветных металлов. Они устойчивы в водных растворах в течение длительного времени. Эти соединения относятся к числу токсичных веществ, и их предельно допустимая концентрация для вод водоемов составляет 1 мкг/л.
С помощью экстракционно-фотометрических методик, используемых для анализа сточных вод [1] или флотационных растворов [3], можно определять концентрации дитиофосфатов в пробах, превышающие 50 мкг/л. Предлагаемая методика позволяет находить общее содержание дитиофосфатов из пробы объемом 250 мл в диапазоне концентраций 1 - 30 мкг/л [2].
Определение дитиофосфатов основано на образовании в растворе дитиофосфата меди (I), экстракции его из кислой среды четыреххлористым углеродом и реэкстракции меди (I) азотной кислотой. Содержание дитиофосфатов в пробе находят по значению атомной абсорбции меди в аликвоте реэкстракта. Минимально определяемая концентрация составляет 1 мкг/л. Мешающих влияний на определение дитиофосфатов с медью (I) атомно-абсорбционным методом не обнаружено.
Для выполнения анализа применяются:
атомно-абсорбционный спектрофотометр с непламенной атомизацией проб любой марки;
весы лабораторные 2-го класса точности с наибольшим пределом взвешивания 200 г по ГОСТ 24104;
центрифуга с набором центрифужных пробирок, например типа ЦЕЛ-1, по ТУ 5-375-4166;
электроплитка с закрытой спиралью мощностью 800 Вт по ТУ 92-208;
баллон газовый для азота (гелия) по ГОСТ 949;
колбы мерные на 100; 250; 500 мл по ГОСТ 1770;
воронки делительные на 500 мл по ГОСТ 25336;
пипетки на 2; 5; 10; 20 мл по ГОСТ 20292;
стаканы на 400 мл по ГОСТ 25336;
цилиндры мерные на 50 мл по ГОСТ 1770;
воронки химические типа В диаметром 50 - 80 мм по ГОСТ 25336;
бюретки с двухходовым краном и автоматическим нулем на 25 мл по ГОСТ 20292;
колбы короткогорлые со шлифом на 250 мл по ГОСТ 25336;
бюксы по ГОСТ 25336;
батометр пластмассовый или стеклянный ГР-18 по ТУ 2504-2507;
фильтры бумажные, тип ФОМ, по ТУ 6-09-1678;
вода дистиллированная по ГОСТ 6709;
бумага лакмусовая нейтральная по ТУ 6-09-3405;
дибутилдитиофосфат натрия технический (аэрофлот натриево-бутиловый) по ТУ 6-02-1073;
медь хлористая, ч., по ГОСТ 4164;
кислота соляная, ч.д.а., по ГОСТ 3118;
кислота азотная, ч.д.а., по ГОСТ 4461;
калия гидроксид, ос.ч., по ОСТ 6-01-301;
эфир диэтиловый технический по ГОСТ 6265-74;
углерод четыреххлористый, ос.ч., по ТУ 6-09-3219 или х.ч. по ТУ 6-09-2663;
натрий хлористый, х.ч., по ГОСТ 4233;
ацетон, ос.ч., по ТУ 6-09-3513;
бензол, х.ч., по ТУ 6-09-779;
спирт этиловый ректификат высший сорт по ГОСТ 18300;
азот особой чистоты по ГОСТ 9293 или поверочный нулевой газ (ПНГ);
бромкрезоловый пурпурный, ч., по ТУ 6-09-1386.
Пробы морской воды для определения дитиофосфатов отбирают чистым пластмассовым или стеклянным батометром. При отборе следует избегать попадания загрязнения в пробу.
Во избежание сорбции дитиофосфатов пробу хранить не рекомендуется. Если окончание анализа (измерение атомной абсорбции) нельзя провести сразу, следует довести первичную обработку пробы до стадии реэкстракции включительно. Устойчивость реэкстрактов сохраняется в течение длительного времени при условии хранения их в чистых склянках с плотными пробками.
4.1.1. Для приготовления раствора хлористой меди концентрацией 0,25 моль/л растворяют 2,55 г хлористой меди в 10 мл концентрированной соляной кислоты и доводят объем раствора дистиллированной водой до 100 мл. Раствор готовят непосредственно перед обработкой проб. Устойчив в течение 6 - 8 ч при хранении в закрытой склянке <*>.
--------------------------------
<*> Кристаллы хлористой меди белого цвета, однако при стоянии на воздухе быстро зеленеют из-за образования основной соли, поэтому для анализа используют реактив зеленого цвета, который дополнительной очистке не подлежит.
4.1.2. Для приготовления спиртового раствора гидроксида калия концентрацией 0,05 моль/л 0,2800 г гидроксида калия растворяют в этиловом спирте и доводят объем раствора до 250 мл.
4.1.3. Раствор бромкрезолового пурпурного 0,1%-ный готовят растворением 0,05 г бромкрезолового пурпурного в 50 мл этилового спирта.
4.1.4. Насыщенный раствор хлористого натрия готовят растворением 36,0 г хлористого натрия в 100 мл дистиллированной воды. Полученный раствор фильтруют.
Перед построением градуировочного графика необходимо провести очистку технического препарата дибутилдитиофосфата натрия, называемого "аэрофлот бутиловый" и содержащего обычно около 60% чистого вещества. Для этого 10 - 15 г дибутилдитиофосфата сначала перекристаллизовывают из этилового спирта, затем растворяют в небольшом количестве горячего ацетона и осаждают из раствора диэтиловым эфиром. Отфильтрованные кристаллы промывают диэтиловым эфиром и сушат на воздухе. Выход очищенного продукта составляет 45 - 50%. Хорошо высушенную соль хранят в бюксе с притертой пробкой. Реактив устойчив в течение нескольких месяцев.
в очищенном дитиофосфате натрия
В стакан емкостью 50 мл помещают 0,2 г очищенного реактива, взвешенного с погрешностью не более 0,0001 г, приливают 10 мл раствора хлористого натрия, 5 мл бензола и 0,3 мл концентрированной соляной кислоты. После перемешивания содержимое стакана количественно переносят в делительную воронку на 50 мл N 1, смывая стенки стакана 15 мл бензола. Смесь в воронке встряхивают 20 раз для извлечения в водный слой неорганических примесей. После полного разделения фаз водный слой сливают в делительную воронку на 50 мл N 2. В воронку N 1, содержащую бензольный слой, приливают еще 10 мл раствора хлористого натрия, повторяют встряхивание и разделение слоев, собирая водный слой в воронку N 2. Операцию встряхивания и разделения повторяют в третий раз, собирая водный слой в воронку N 2. Бензольный слой в воронке N 1, содержащий дибутилдитиофосфорную кислоту, сохраняют. К объединенному в воронке N 2 промывному раствору приливают 10 мл бензола, встряхивают 20 раз для окончательного извлечения в бензольный слой следов дибутилдитиофосфорной кислоты. Нижний водный слой отбрасывают.
Верхний бензольный слой сливают в коническую колбу объемом 100 мл, смывая стенки воронки 5 мл бензола. В эту же колбу количественно переносят бензольный слой из воронки N 1, также промывая ее 5 мл бензола. К объединенному бензольному экстракту добавляют 2 мл спирта, 35 - 40 мл воды и пять капель индикатора бромкрезолового пурпурного, затем титруют раствором гидроксида калия до появления в водном растворе фиолетовой окраски.
Содержание (%) дибутилдитиофосфата натрия в процентах рассчитывают по формуле
 ,
,где V - объем спиртового раствора гидроксида калия концентрацией 0,05 моль/л, израсходованный на титрование, мл; 0,0132 - количество дибутилдитиофосфата натрия, соответствующее 1 мл точно спиртового раствора гидроксида калия концентрацией 0,05 моль/л; m - навеска очищенного реактива, г.
За результат анализа принимают среднее арифметическое двух параллельных определений, допускаемое расхождение между которыми не должно превышать 0,5%.
Содержание основного вещества в дважды очищенном техническом продукте составляет обычно около 95%.
Методика определения дитиофосфатов требует большой тщательности выполнения. Особое внимание следует уделить склянкам для сбора экстрактов. Необходимо проводить строгий контроль их чистоты. Для этого предварительно вымытую склянку (коническую колбочку со шлифом) ополаскивают 3 - 5 мл разбавленной азотной или соляной кислоты, после чего промывную кислоту анализируют на атомно-абсорбционном спектрофотометре. Если абсорбция превышает значение 0,010, то склянку замачивают в разбавленной кислоте на несколько часов, затем ополаскивают несколько раз и снова проверяют на чистоту. При необходимости процедуру очистки повторяют до тех пор, пока абсорбция не станет меньше указанного значения.
Пробу объемом 250 мл переносят в делительную воронку на 500 мл, добавляют 3 мл раствора хлористой меди и 1 мл концентрированной соляной кислоты (проба должна быть подкислена до pH = 1...2). После перемешивания раствора добавляют в воронку 5 мл четыреххлористого углерода и экстрагируют в течение 3 мин. После разделения слоев (через 7 - 10 мин) нижний органический слой собирают в склянку емкостью 15 - 20 мл, фильтруя его через кусочек ваты, предварительно вымытой растворителем и высушенной. Если после экстракции образовалась эмульсия, то ее собирают в центрифужную пробирку и центрифугируют 10 мин при скорости 3000 об./мин. Содержимое пробирки вновь возвращают в делительную воронку и собирают отделившийся экстракт, как указано выше. Необходимо строго следить, чтобы ни одна капля водной фазы не попала в склянку. Склянки должны быть чистыми и сухими. Затем добавляют к экстракту 200 мкл концентрированной азотной кислоты, закрывают пробкой и встряхивают в течение 30 с, после чего добавляют еще 2 мл дистиллированной воды и снова встряхивают. После разделения слоев верхний водный слой (реэкстракт) подготовлен к измерению.
Для выполнения холостого определения 250 мл дистиллированной воды проводят через все стадии анализа, предусмотренные для пробы. При соблюдении всех требований, предъявляемых к чистоте посуды и реактивов, значения атомной абсорбции холостых определений не превышают обычно 0,010 - 0,015. Холостое определение проводят перед построением градуировочного графика и повторяют для каждой новой партии реактивов.
Основной стандартный раствор дибутилдитиофосфата натрия готовят, растворяя навеску около 0,5 г (с учетом найденного содержания основного вещества) в дистиллированной воде и доводя объем раствора до 100 мл дистиллированной водой. Концентрация дибутилдитиофосфата натрия в основном стандартном растворе 5 г/л. Раствор устойчив.
Расчет необходимой навески m (г) проводят по формуле
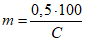 ,
,где C - найденное содержание основного вещества, %.
Например, если содержание основного вещества в очищенном препарате - 95%, то навеска составляет 0,5263 г.
Для приготовления рабочего стандартного раствора дибутилдитиофосфата натрия 0,25 мл основного раствора помещают в мерную колбу на 500 мл и доводят объем до метки дистиллированной водой. Концентрация дибутилдитиофосфата натрия в рабочем стандартном растворе 2,5 мг/л. Раствор готовят в день построения градуировочного графика.
Для приготовления градуировочных растворов в ряд мерных колб объемом 250 мл вносят 0,1; 0,5; 1,0; 1,5; 2,0; 3,0 мл рабочего стандартного раствора дитиофосфата и доводят объемы растворов до метки дистиллированной водой. Полученные концентрации составляют соответственно 1; 5; 10; 15; 20; 30 мкг/л. Обработку каждого градуировочного раствора производят так же, как обработку пробы при проведении анализа. Каждый градуировочный раствор готовят параллельно не менее трех раз.
Градуировочный график строят по средним измеренным значениям атомной абсорбции в координатах "атомная абсорбция - концентрация дитиофосфатов в пробе", при этом из каждого значения атомной абсорбции градуировочного раствора вычитают значение для холостой пробы.
спектрофотометра
Рабочие параметры и температурно-временной режим атомизации проб устанавливают в соответствии с инструкцией прибора.
После разделения слоев отбирают из верхнего водного слоя 20 мкл с помощью пипетки Эппендорфа и вводят в камеру атомизации атомно-абсорбционного спектрофотометра. Интенсивность абсорбции меди измеряют при 324,7 нм, используя следующий температурно-временной режим работы:
сушка при 80 °С - 30 с;
обжиг при 900 °С - 20 с;
атомизация при 2500 °С - 10 с.
По найденному значению атомной абсорбции с помощью градуировочного графика находят концентрацию дитиофосфатов в пробе (мкг/л), при этом из каждого значения атомной абсорбции необходимо вычесть величину холостого определения.
На основании метрологической аттестации, проведенной ВНИИАСМ-НПО "Исари" Госстандарта СССР с 01.09 по 20.12.89 (табл. 49), настоящая методика определения дитиофосфатов допущена к применению в организациях Росгидромета.
Таблица 49
Результаты метрологической аттестации
Диапазон концентрации дитиофосфатов в морской воде, мкг/л | Показатель воспроизводимости | Показатель правильности | Показатель погрешности МВИ, суммарная погрешность |
1,0 - 10,0 | 12,0 | 31,1 | 34,4 |
10,1 - 30,0 | 3,4 | 8,2 | 9,1 |
К выполнению анализа допускаются лица с высшим или средним специальным образованием, имеющие опыт работы с химическими препаратами и на атомно-абсорбционном спектрофотометре, допущенные к работе с газовыми баллонами.
Для анализа 10 проб требуется 12,3 чел.-ч, в том числе:
на взятие проб из батометра - 0,3 чел.-ч;
на приготовление растворов реактивов - 1 чел.-ч;
на подготовку посуды - 3 чел.-ч;
на подготовку атомно-абсорбционного спектрофотометра к работе - 1,5 чел.-ч;
на проведение экстракции - 4 чел.-ч;
на выполнение измерений - 2,5 чел.-ч.
2. Определение дитиофосфатов в морской воде. Методические указания. РД 52.10.240-90. М.: Госкомгидромет, 1990. 15 с.
3. M. Jones, J. Woodcock. Determination of diethyldithiophosphate in flotation liquors by solvent extraction and ultraviolet spectrometry. Anal. Chem., 1986, v. 58, N 8, p. 1845 - 1848.
Нефтепродукты (НП), попадающие в моря и океаны с эксплуатационными сбросами судов (балластные, льяльные воды, потери при грузовых операциях и т.п.), составляют существенную часть суммарной массы нефтяного загрязнения - около 40%. Борьба с виновниками эксплуатационных сбросов объективно возможна только с помощью законоположений о санкциях за загрязнение моря. Общепризнанной правовой основой санкций служат специально разработанные и постоянно усовершенствуемые системы идентификации нефтяных разливов (СИ), включающие в себя ряд химико-аналитических методик. Ниже описывается СИ, состоящая из минимального числа (трех) методик и процедуры искусственного выветривания (старения) образцов. Система предусматривает физико-химическое исследование загрязняющих море НП наряду с анализом НП, взятых от возможных источников этого загрязнения (судов), с целью идентификации этих продуктов. Вероятность установления соответствия составляет около 0,9.
Другие известные СИ [1 - 3] перегружены методиками, среди которых имеются как очень сложные и поэтому малодоступные, так и простейшие, но малоинформативные; существенным недостатком этих СИ является неучет или же лишь теоретический учет эффектов выветривания НП.
Система предназначена для использования в исследовательских лабораториях Минэкологии, привлекаемых к работе по выявлению виновников нефтяного загрязнения морских акваторий в случаях эксплуатационных разливов, а также по установлению источников разливов НП в аварийных ситуациях. Кроме того, СИ может быть применена для физико-химической идентификации сликов (пятен) НП на морских акваториях при наблюдениях за эволюциями аварийных и искусственных разливов, обусловленными дрейфом, рассеиванием, диспергированием, агрегацией и другими факторами.
В СИ включены три методики исследования НП, основанные на методах спектрофлуорометрии (СФ), жидкостной хроматографии высокого давления (ЖХВД) и капиллярной газовой хроматографии (КГХ), при этом первые две методики служат для скрининга (предварительного анализа типа НП), а третья - для идентификации.
Светлые (не содержащие смол и пигментов) образцы НП анализируют всеми тремя методами непосредственно, тогда как окрашенные образцы - методами СФ и КГХ, а для анализа методом ЖХВД берут образцы НП, осветленные способом адсорбционной хроматографии на оксиде алюминия и силикагеле.
Реперные НП от возможных источников загрязнения, подвижных и стационарных, подвергают искусственному старению (выветриванию) для увеличения корректности сравнительных анализов, воздействуя водой, ультрафиолетовым облучением (УФ), искусственным ветром. Из всех образцов НП перед их исследованием удаляют низкокипящие компоненты отгонкой с н-гептаном.
Идентификация сводится к сравнению характеристик НП из разлива (слика, пятна) и реперных НП по соответствующим флуороспектрограммам и хроматограммам с помощью визуальных и математических критериев.
Минимальная необходимая для анализа масса вещества по описываемой СИ составляет 1 г.
Для выполнения анализов применяются:
спектрофлуорометр с разверткой длин волн возбуждения и эмиссии в интервале 250 - 550 нм любой марки;
хроматограф жидкостной высокого давления любой марки с колонкой из нержавеющей стали длиной 60 - 200 мм с внутренним диаметром 2 - 6 мм, наполненной силасорбом 600 (прямая фаза) или силасорбом C18 (обращенная фаза) зернением 5 - 7,5 мкм;
хроматограф газовый с программатором температуры и пламенно-ионизационным детектором, предназначенный для работы с капиллярной колонкой, скомплектованный с капиллярной колонкой из плавленого кварца длиной 25 - 50 м с внутренним диаметром 0,25 - 0,4 мм с адгезионно или химически связанной пленкой НЖФ (группы полисилоксана);
электроплитка с закрытой спиралью мощностью 250 Вт и более по ТУ 92-208;
баллоны газовые для азота, водорода, воздуха по ГОСТ 949;
редукторы кислородный и водородный по ГОСТ 6268;
вентилятор электрический любой марки, например типа ВО-45, по ГОСТ 7402;
облучатель ртутно-кварцевый ОРК-21 по ТУ 64-1-1618;
устройство для отбора НП с поверхности моря - пластина лиофильного материала на держателе, соединенном с линем;
микрошприц вместимостью 10 мкл любой марки, например МШ-10;
пинцет по ТУ 2-31-32;
пипетка стеклянная длиной 320 мм, диаметром 6 мм с резервуаром диаметром 25 мм с боковым отводом (рис. 27);

1 - силикагель; 2 - оксид алюминия; 3 - стекловата
аппарат для перегонки на шлифах, включающий в себя колбу типа "0" (куб) вместимостью 25 мл; насадку с одной горловиной типа Н1 14/23; холодильник типа ХПТ-1 14/23; пробирку с конусом (приемник) типа П4 вместимостью 25 мл - все детали по ГОСТ 25336;
воронка лабораторная типа "В" диаметром 36 мм по ГОСТ 25336;
пробирки мерные типа ПГКШ вместимостью 5 или 10 мл по ГОСТ 10515;
склянки с притертыми пробками по ТУ 6-19-6 или колбы типа Кн-1 по ГОСТ 25336;
трубка U-образная ТХ 45° - 14/23 по ГОСТ 25336 с активным углем;
склянка СПЖ по ГОСТ 25336 (склянка Тищенко);
склянка СПТ по ГОСТ 25336 ("сухая" склянка Тищенко);
стакан стеклянный вместимостью 250 или 500 мл по ГОСТ 8682;
чашка фарфоровая выпарительная вместимостью 150 мл по ГОСТ 9147;
вата стеклянная;
эксикатор диаметром 230 мм или более, например типа II, по ГОСТ 25336;
азот газообразный, ос.ч., по ГОСТ 9293 или гелий газообразный технический по МРТУ 51-940;
алюминия оксид, ч., для хроматографии по ТУ 6-09-3916;
силикагель марки АСК по ГОСТ 3956;
уголь активный, БАУ по ГОСТ 6217;
пентан, ч., по ТУ 6-09-3661;
эфир этиловый (серный) для наркоза по Госфармакопее Х, ст. 35;
н-гексан, ч., по ТУ 6-09-3375;
н-гептан, ч., по ГОСТ 5395;
натрий сернокислый безводный по ГОСТ 4166;
натрий хлористый, х.ч., по ГОСТ 4233;
стандартные вещества:
н-гексадекан, ч., по ТУ 6-09-3660;
октадекан, ч., по ТУ 6-09-3005;
трикозан, ч., по ТУ 6-09-1842;
эйкозан, ч., по ТУ 6-09-1842.
Пробы НП из разлива на поверхности моря в зависимости от толщины слоя d отбирают: экранным устройством (сетка из нержавеющей стали с ячейками до 1 мм2 на раме), например 25 х 25 см - при d <= 1 мм; устройством (см. п. 2) с пластинами из поглощающих или адгезирующих НП материалов: поролона, тефлона и т.п. - при d = 50...1 мм; способом черпания - при d = 0,5 см и более. Все средства отбора должны быть соответствующим образом подготовлены: промыты гексаном и высушены. Хранить их после этой процедуры следует в чистых полиэтиленовых мешках, предварительно завернутыми в бумагу. Трос (линь), к которому прикрепляют устройства, не должен быть загрязнен каким-либо посторонним НП. При использовании сетчатого пробоотборника смывают с него НП окунанием в гептан, налитый в металлическую ванночку или кювету достаточных размеров. НП с пластиночных устройств можно переносить в такую же ванночку без применения растворителя (образец соскабливают шпателем) либо с помощью гептана. В случае простого черпания НП с поверхности моря использовать ванночку необязательно.
Реперные НП от возможных источников отбирают каким-либо из доступных методов, например из числа описанных выше, как правило, без употребления растворителя.
В среднем масса НП для анализов должна составлять 1 - 5 г.
Подлежащие идентификации образцы НП или их гептановые растворы помещают в склянки (колбы) с притертыми пробками, удаляют воду пипеткой Пастера (трубка с оттянутым концом). Хотя в закупоренном виде под слоем инертного газа образцы могут сохраняться в холодильнике около полумесяца в неизменном виде, их исследование рекомендуется проводить как можно быстрее по окончании отбора.
4.1.1. Оксид алюминия II степени активности готовят дезактивацией добавкой 3% дистиллированной воды к прокаленному при 350 - 400 °С в течение 6 ч реактиву.
4.1.2. Силикагель измельчают до зернистости 40 - 100 мкм и нагревают в сушильном шкафу 6 ч при 250 °С.
4.1.3. Активный уголь высушивают 2 ч при 100 - 110 °С.
Гептановые растворы НП (образцы, отобранные без применения растворителей, растворяют в гептане; концентрация раствора должна быть около 10 мг/мл) высушивают безводным сернокислым натрием, переносят в перегонный аппарат. В горловину насадки вставляют трубку с оттянутым в капилляр концом, касающимся дна колбы, подсоединяют трубку через редуктор к баллону с азотом, гелием, аргоном или углекислым газом. Вместо баллона допускается пользоваться резиновой камерой по ТУ 38-10-6179, наполненной одним из указанных газов. Между редуктором (или камерой) и трубкой устанавливают кран, U-образную трубку с активным углем и склянку Тищенко с нелетучей жидкостью, например с глицерином. Под куб подставляют баню с глицерином или силиконовой жидкостью; температуру в бане, нагреваемой электроплиткой, рекомендуется поддерживать посредством электрореле и контактного термометра. Отгонку гептана ведут в токе газа со скоростью около 5 мл/мин при температуре бани 120 °С. Кубовый остаток упаривают в фарфоровой чашке на водяной бане при 80 - 90 °С в течение 10 мин. В итоге этой процедуры получают термообработанный НП.
Сильноокрашенные образцы (нефти, мазуты, гудроны и т.п.) для ЖХВД анализа осветляют сорбцией на оксиде алюминия и силикагеле. Процедуру, цель которой заключается в предохранении колонки хроматографа от загрязнения, производят следующим образом. В стеклянную пипетку (см. рис. 27) насыпают оксид алюминия слоем 10 см (около 2 г), затем - силикагель слоем 2 см (около 0,3 г). Боковой отвод пипетки присоединяют через кран к источнику сжатого воздуха, в качестве которого можно использовать микрокомпрессор или наполненную воздухом резиновую камеру; между источником и пипеткой целесообразно сделать ответвление с краном для регулировки давления воздуха в пипетке.
В пипетку наливают смесь пентан - эфир 9:2, закрывают отверстие пробкой, открывают кран и дают растворителю впитаться в сорбент, а избытку стечь. Затем, закрыв кран, вносят в пипетку около 10 мг НП - как такового либо в виде раствора в небольшом количестве гексана. Создают в пипетке давление и дают пробе впитаться в сорбент, после чего хроматографируют сорбат той же смесью пентана и эфира, отбирая 6 - 8 мл элюата. Упариванием последнего получают "сухой остаток" пробы. Процедуру проводят 2 - 3 раза, получая таким образом три пробы каждого образца НП.
Реперные НП от возможных источников перед термообработкой дополнительно подвергают искусственному выветриванию. Процедура заключается в следующем. 1 - 3 г НП помещают на поверхность природной либо искусственной морской воды (30 г поваренной соли на 1 л дистиллированной воды), налитой доверху в эксикатор, включают направленные на образец источник УФ-излучения (ОРК-21), вентилятор и проводят выветривание в течение 4 ч. По окончании процедуры НП переносят с помощью чистого поролонового тампона, зажатого в пинцете, в склянку (колбу), растворяют в гептане и высушивают безводным сульфатом натрия.
Спектрофлуорометрическому скринингу подвергают непосредственно любые образцы, подготовленные согласно п. 4.2; скринингу методом ЖХВД - только бесцветные или слабоокрашенные НП (например, топлива) либо осветленные образцы; идентификации по методу КГХ - образцы любого типа.
Сравниваемые образцы НП (из разлива и реперный) анализируют в первую очередь методом СФ. В случае выявленного несоответствия образцов (критерии см. п. 8.1) данный реперный образец исключают из рассмотрения и берут другой возможный реперный НП. В случае соответствия сравниваемых НП или при получении неопределенного результата их подвергают скринингу методом ЖХВД, при этом окрашенные НП предварительно осветляют. Результаты скрининга ЖХВД должны подтверждать данные СФ-скрининга. Однако в силу практической недостижимости адекватного выветривания образцов эти результаты могут и расходиться. Критерии соотносимости (соответствия или несоответствия) образцов НП по данным ЖХВД приведены в п. 8.2. Приоритетным следует считать скрининг методом СФ, так как его результаты менее подвержены влиянию степени выветренности анализируемых образцов. Анализ методом КГХ является обязательным этапом идентификации; это означает, что он производится во всех случаях, когда СФ-скринингом установлено соответствие сравниваемых образцов. Критерии соотнесения НП методом КГХ приведены в п. 8.3.
Подключение к сети, проверку работоспособности, профилактику приборов следует проводить в соответствии с инструкциями по их эксплуатации.
После того как прибор выйдет на режим, устанавливают рабочие условия:  = 254 и 310 нм; развертка
= 254 и 310 нм; развертка  = 250...550 нм со скоростью 25 нм/мин; чувствительность средняя; постоянная времени 1 с; ширина щелей монохроматоров 5 нм; скорость диаграммной ленты 1 - 2 см/мин. Для измерений служат прямоугольные кварцевые кюветы 1 х 1 х 4,5 см. Концентрация аналитических растворов (АР) около 20 мкг/мл.
= 250...550 нм со скоростью 25 нм/мин; чувствительность средняя; постоянная времени 1 с; ширина щелей монохроматоров 5 нм; скорость диаграммной ленты 1 - 2 см/мин. Для измерений служат прямоугольные кварцевые кюветы 1 х 1 х 4,5 см. Концентрация аналитических растворов (АР) около 20 мкг/мл.
Для поверки прибора, которая должна производиться не реже одного раза в год, а при необходимости чаще, рекомендуется использовать 3,4-бензпирен. Снимаются спектры возбуждения при  = 405 нм и спектры эмиссии при
= 405 нм и спектры эмиссии при  = 295 нм раствора этого ПАУ в циклогексане или бензоле, концентрация 100 нг/мл. Получаемые спектрофлуорограммы должны в основном соответствовать прилагаемым к прибору спектрам по высоте и положению пиков при идентичных рабочих условиях.
= 295 нм раствора этого ПАУ в циклогексане или бензоле, концентрация 100 нг/мл. Получаемые спектрофлуорограммы должны в основном соответствовать прилагаемым к прибору спектрам по высоте и положению пиков при идентичных рабочих условиях.
По выходе прибора на режим устанавливают рабочие условия: расход элюента (гексана в случае силасорба 600 и этанол-аммиак-вода 8:1:1 в случае обращенной фазы) 0,1 - 0,5 мл/мин; чувствительность средняя;  - 210 и 254 нм; объем проб - 0,8...5 мкл; скорость самописца - 1 см/мин. Концентрация АР - около 10 мг/мл.
- 210 и 254 нм; объем проб - 0,8...5 мкл; скорость самописца - 1 см/мин. Концентрация АР - около 10 мг/мл.
Работоспособность жидкостного хроматографа проверяют один раз в год или чаще (например, при смене колонки) по контрольным растворам аренов, например, о-, м- и п-нитроанилинов по прилагаемой к прибору инструкции. Параметры контрольных хроматограмм (коэффициент разделения, число теоретических тарелок, высота эквивалентной теоретической тарелки) должны соответствовать паспортным данным прибора в пределах +/- 20%. В противном случае прибор подлежит техническому освидетельствованию.
Капиллярную колонку с жидкой фазой подсоединяют к испарителю и детектору хроматографа в соответствии с инструкцией по эксплуатации прибора. Об окончании кондиционирования судят по отсутствию или неизменности уровня шумов и дрейфа нулевой линии самописца при рабочей температуре и выбранной чувствительности.
Условия хроматографирования: скорость газа-носителя (гелия, азота) на выходе из колонки - 20...30 см/с; скорость подачи водорода - 30, воздуха - 300 мл/мин; температура испарителя - 280...300 °С, детектора - 350 °С, начальная температура термостата - 40...80 °С; температурная программа: изотерма 1,5 - 3 мин при начальной температуре, далее - линейное программирование с шагом 4 - 6 °С до конечной температуры 270 - 300 °С, после чего - изотермический режим до полного выхода компонентов (около 30 мин); чувствительность средняя; объем пробы, вводимой шприцем, - 0,5...1,5 мкл; делитель потока 1:50; скорость ленты самописца - 1 см/мин.
Один раз в год или при необходимости чаще хроматограф поверяется по контрольным растворам н-алканов, например C16 - C23. Если при этом коэффициенты разделения, число теоретических тарелок и высота эквивалентной теоретической тарелки воспроизводятся в пределах +/- 20%, то прибор пригоден к работе.
Чистую и сухую кювету наполняют АР термообработанного НП с концентрацией около 20 мкг/мл и, убедившись в чистоте внешних поверхностей, устанавливают в кюветодержатель подготовленного к работе спектрофлуорометра. При  = 254 нм сканируют
= 254 нм сканируют  вручную по всему интервалу 250 - 500 нм, следя за отклонением пера самописца при выключенной диаграммной ленте. Найдя
вручную по всему интервалу 250 - 500 нм, следя за отклонением пера самописца при выключенной диаграммной ленте. Найдя 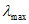 флуоресценции, устанавливают это значение приблизительно на уровне 80% ширины ленты, варьируя чувствительность. Закрывают шторку, жидкость в кювете заменяют новой порцией того же АР, монохроматор эмиссии устанавливают на начальное значение 250 нм. Повторно сканируют
флуоресценции, устанавливают это значение приблизительно на уровне 80% ширины ленты, варьируя чувствительность. Закрывают шторку, жидкость в кювете заменяют новой порцией того же АР, монохроматор эмиссии устанавливают на начальное значение 250 нм. Повторно сканируют  в автоматическом режиме, предварительно включив диаграммную ленту. Записывают спектр (типичная спектрофлуорограмма нефти приведена на рис. 28а). Флуорометрирование образцов производят по 3 раза, причем каждый раз с новой порцией АР. Затем устанавливают
в автоматическом режиме, предварительно включив диаграммную ленту. Записывают спектр (типичная спектрофлуорограмма нефти приведена на рис. 28а). Флуорометрирование образцов производят по 3 раза, причем каждый раз с новой порцией АР. Затем устанавливают  = 310 нм и повторяют всю вышеописанную работу, получают спектрофлуорограмму (рис. 28б, прибор FP-550 фирмы "Джаско", Япония; чувствительность - X1/10, постоянная времени - 1 с).
= 310 нм и повторяют всю вышеописанную работу, получают спектрофлуорограмму (рис. 28б, прибор FP-550 фирмы "Джаско", Япония; чувствительность - X1/10, постоянная времени - 1 с).
на спектрофлуорометре FP-550 "Джаско", Япония
а)  - 254 нм; б)
- 254 нм; б)  - 254 нм
- 254 нм
Аналитические растворы готовят растворением "сухих остатков", осветленных НП, получаемых согласно п. 4.3, в 0,5 - 1,0 мл гексана.
Заполняют насос вышедшего на режим жидкостного хроматографа элюентом, устанавливают скорость подачи 0,1 - 0,5 мл/мин. С помощью устройства ввода проб вводят в колонку заданный объем АР в интервале 0,8 - 5,0 мкл, затем включают насос, ленту самописца, отметчик объема элюата. Поступая согласно инструкции к прибору, начинают хроматографирование образца, которое ведут до выхода всех компонентов его состава. Хроматографирование каждого "сухого остатка" проводят 2 - 3 раза. Типичная хроматограмма дизельного топлива представлена на рис. 29 (прибор "Милихром-1", колонка 64 х 2 мм с силасорбом 600 зернением 5 мкм; чувствительность 0,60; постоянная времени 0,15 с; расход элюента гексана 0,1 мл/мин;  - 210 нм).
- 210 нм).

полученная на жидкостном хроматографе "Милихром-1"
Вводят в испаритель готового к работе хроматографа 1 мкл градуировочного раствора н-алканов C16 - C23, приготовленного растворением 5 - 15 мг алканов в 5 мл гексана и разбавлением раствора приблизительно в 100 раз. Одновременно с вводом пробы запускают программу анализа, по окончании которого получают хроматограмму. Значения времени удерживания н-алканов должны прямо пропорционально зависеть от числа атомов C в углеводородах. Эту градуировочную хроматограмму в дальнейшем используют для соотнесения пиков на хроматограммах исследуемых НП из разлива и от источника.
Анализируют либо термообработанные образцы НП, либо "сухие остатки" осветленных НП в виде растворов, например в гексане, хлороформе, концентрацией 0,1 - 5 мг/мл. Вводят в хроматограф около 1 мкл АР и одновременно запускают программу. По окончании программы получают хроматограммы вида, изображенного на рис. 30 (прибор "Vista 6000" фирмы Вариан, США). Хроматографирование каждого образца производят 2 - 3 раза, а одного из них с наиболее полным набором н-алканов (контрольный образец) - 5 - 8 раз для установления показателя воспроизводимости дельта результатов хроматографического анализа.

нефтепродукта из разлива, полученная на хроматографе
VISTA 6000 "Вариан" (США)
8.1.1. Спектрофлуорограммы НП (см. рис. 28) характеризуют: 1) контуром (общей формой); 2) лямбда максимумов эмиссии; 3) числом максимумов и плеч; 4) отношением р высот при максимумах (средним арифметическим по трем спектрам). Если совпадают первые три параметра сравниваемых спектрограмм и соизмеримы р - в пределах +/- 10%, то НП в первом приближении считают соответствующими друг другу. При несовпадении всех четырех параметров НП считают несоответствующими. В промежуточных случаях результаты полагают неопределенными.
8.1.2. Жидкостные хроматограммы (см. рис. 29) характеризуют:
1) контуром; 2) числом пиков ("веществ"); 3) временами выхода (объемами удерживания) "веществ"; 4) отношением высот пиков. При совпадении двух первых характеристик и соизмеримости в пределах +/- 10 - 15% средних арифметических значений 3-го и 4-го параметров считают, что сравниваемые НП соответствуют друг другу по типу. При несовпадении параметров НП полагают несоответствующими.
8.1.3. На типичных капиллярных газовых хроматограммах (см. рис. 30) видны сигналы (линии) н- и изоалканов ("забор") на выпуклом фоне совокупности неподеленных компонентов ("горбе"). В большинстве случаев контуры "забора" и "горба" специфичны, поэтому при сильном различии контуров можно констатировать несоответствие характеризуемых ими образцов. В отсутствии явно выраженного несходства внешнего вида хроматограммы подвергают детальному исследованию. Последнее заключается в следующем.
На двух-трех хроматограммах одного и того же АР находят идентичные сигналы и определяют их интенсивности (высоты пиков) h с точностью 0,5 мм, отсчитывая от истинной нулевой линии (рис. 30). Вычисляют среднеарифметические значения h. Аналогично определяют средние высоты пиков H реперного НП. Затем находят отношения высот идентичных пиков (с одинаковым временем удерживания) h1/H1, h2/H2, h3/ H3 ... и т.д. НП считают соответствующими друг другу, если не менее 75% отношений соизмеримы с точностью +/- 10%. Отсутствие единичных пиков или, напротив, их наличие сверх пиков хроматограммы репера необязательно свидетельствует о несоответствии сравниваемых НП.
Хотя при сравнительном исследовании нет необходимости знать, каким конкретным соединениям (углеводородам) принадлежат те или иные пики, и идентификацию НП можно с успехом проводить "вслепую", т.е. так, как описано выше, иногда рационально оперировать "именными отношениями" (например, при представлении результатов интеркалибраций). Расшифровку пиков осуществляют на основе хроматограмм градуировочных смесей стандартных соединений - н-алканов C16 - C23. Хроматографирование этих смесей производят строго в условиях, принятых в работе по идентификации разливов (см. п. 7.3). Соотнесения в хроматограммах пиков других алканов, не представленных в искусственной смеси, устанавливают, используя закон неизменности разностей времен удерживания любой пары н-алканов CxH2x+2 при линейном программировании температуры.
Зная, каким алканам соответствуют пики на хроматограммах сравниваемых НП, и определив высоты этих пиков, вычисляют отношения  : C16/C17, C17/C18, C18/C19, C19/C20 и т.д., а также C17/пристан и C18/фитан. Кроме приведенных, можно использовать и другие
: C16/C17, C17/C18, C18/C19, C19/C20 и т.д., а также C17/пристан и C18/фитан. Кроме приведенных, можно использовать и другие  : например изо-C18/пристан, фитан/пристан и т.д <*>.
: например изо-C18/пристан, фитан/пристан и т.д <*>.
--------------------------------
<*> Соединения изо-C18H38, пристан (изо-C19H40) и фитан (изо-C20H42) - реликтовые углеводороды нефти изопреноидного строения, которые подлежат обязательному рассмотрению при идентификации НП, выходят после н-алканов C16, C17 и C18 соответственно.
Далее вычисляют относительное расхождение P:
 ,
,где  - отношение для НП из разлива;
- отношение для НП из разлива;  - отношение для реперного НП (от возможного источника);
- отношение для реперного НП (от возможного источника); 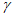 - число отношений.
- число отношений.
Если P <= 15, то образцы соответствуют; если P > 15, то не соответствуют. Это условие выполняется в том случае, когда относительная погрешность расчета  (показатель воспроизводимости
(показатель воспроизводимости  ) для контрольного образца с вероятностью 0,95 не превышает 10%.
) для контрольного образца с вероятностью 0,95 не превышает 10%.
Вывод о соотнесении сравниваемых образцов НП из разлива и от возможного источника делается на основе комплексного сопоставления результатов анализов (скрининга и идентификации) в соответствии с табл. 50.
Таблица 50
Варианты соотнесения образцов НП
Тип сравниваемой пары НП | Результаты скрининга | Результат идентификации | Вывод | |
СФ | ЖХВД | |||
I | + | + или +/- | + | + |
II | + | - или +/- | + | + |
III | + | - | - | - |
IV | - | 0 | 0 | - |
Примечание. "+" - образцы пары НП соответствуют; "-" - образцы пары НП не соответствуют; "+/-" - результат неопределенный; "0" - исследование не проводят.
При проведении работы по идентификации НП согласно настоящей СИ могут быть случаи, когда вполне определенный вывод сделать невозможно. В таких случаях рекомендуется применять иные методы скрининга (например, по содержанию никеля и ванадия) и идентификации (например, хроматомасс-спектрометрию), описанные в других СИ.
Работу по идентификации могут выполнять опытные инженер-химик и техник-химик при консультативной помощи научных сотрудников - специалистов по спектрометрии и хроматографии.
На проведение исследования 10 образцов НП требуется 75 чел.-ч, в том числе:
на взятие проб - 0,5 чел.-ч;
на подготовку посуды - 3 чел.-ч;
на сборку установок - 2 чел.-ч;
на термообработку - 20 чел.-ч;
на работу, связанную с выветриванием образцов, - 1 чел.-ч;
на осветление образцов - 12 чел.-ч;
на подготовку средств измерений к работе - 4,5 чел.-ч;
на выполнение измерений:
скрининг методом СФ - 4 чел.-ч;
скрининг методом ЖХВД - 4 чел.-ч;
КГХ идентификация - 15 чел.-ч;
на обработку результатов:
метод СФ - 1,5 чел.-ч;
метод ЖХВД - 2,5 чел.-ч;
метод КГХ - 5 чел.-ч.
2. Identification of Oil pollution. Method of DHI, MERC. 211. Inf. 8. 1985. 19 pp.
3. Nordtest method. Oil spills at sea: Identification. NT Chem. 001. H., Finland, 1983, 34 pp.
ФЕНОЛОВ, ХЛОРИРОВАННЫХ УГЛЕВОДОРОДОВ И ГЕРБИЦИДОВ
Расчет параметров колонки и детектора дан на примере анализа фенолов.
1. Для определения газохроматографических параметров колонок и детекторов несколько раз вводят в испаритель хроматографа по 1 мкл стандартных растворов ацетилпроизводных хлор- и нитрофенолов и алкилфенолов и рассчитывают:
число N теоретических тарелок колонок;
высоту H, эквивалентную теоретической тарелке;
порог чувствительности и линейный диапазон детекторов.
Параметры N и H характеризуют эффективность колонок (N >= 1000, H <= 1 мм).
2. Число теоретических тарелок N рассчитывается по формуле
 ,
,где lR - расстояние на хроматограмме от стартовой отметки до вершины пика, мм; Wb - ширина пика в основании, мм.
Число теоретических тарелок колонки (см. п. п. 6.5, 6.7), используемой для анализа хлор- и нитрофенолов:
 ,
, .
.3. Высота, эквивалентная теоретической тарелке, определяется по формуле
 ,
,где L - длина колонки, мм;
т.е. в случае анализа хлор- и нитрофенолов H = 1800/1710 = 1,05 мм, а в случае алкилфенолов H = 1800/1730 = 1,04 мм.
4. Чувствительность детектора определяется по формуле
 ,
,где S - площадь пика, см2; C - шкала электрометра, на которой велась регистрация сигналов детектора, A; l - длина шкалы самописца, см (как правило, l = 25 см);  - скорость движения диаграммной ленты, см/с; q - количество вещества, вводимого в испаритель хроматографа, мг.
- скорость движения диаграммной ленты, см/с; q - количество вещества, вводимого в испаритель хроматографа, мг.
Пример 1. Для пентахлорфенола: S = 9,48 см2; C = 50·10-12 A;  = 6,67·10-3 см/с; l = 25 см; q = 4,8·10-5 мг. Отсюда
= 6,67·10-3 см/с; l = 25 см; q = 4,8·10-5 мг. Отсюда
 .
.Пример 2. Для 3,4-диметилфенола: S = 0,98 см2; C = 8·10-12 A;  = 5·10-3 см/с; l = 25 см; q = 43,2·10-6 мг. Отсюда
= 5·10-3 см/с; l = 25 см; q = 43,2·10-6 мг. Отсюда
 .
.5. Порог чувствительности Cmin определяют по формуле
 ,
,где сигма - допустимое отклонение, составляющее 5% самой чувствительной рабочей шкалы электрометра и равное 2,5·10-13 А.
При работе с детектором ДЭЗ (пентахлорфенол):
 ,
,а с детектором ПИД (3,4-диметилфенол):
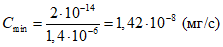 .
.ИЗМЕРЕНИЯ СОДЕРЖАНИЯ ХЛОРИРОВАННЫХ УГЛЕВОДОРОДОВ
НА ГАЗОВОМ ХРОМАТОГРАФЕ С КАПИЛЛЯРНОЙ КОЛОНКОЙ
Капиллярные колонки обладают значительно большей эффективностью по сравнению с набивными колонками при прочих равных условиях и, следовательно, на них можно добиваться повышения качества разделения. В частном случае измерения содержания хлорированных углеводородов в морской воде отмеченное преимущество капиллярных колонок позволяет исключить продолжительную стадию химической обработки - дегидрохлорирования (см. гл. "Хлорированные углеводороды", п. п. 4; 5; 6; 7) и упростить обработку результатов (см. п. 8).
Измерение можно проводить на хроматографе любой марки с детектором электронного захвата, снабженном капиллярной колонкой из стекла длиной 25 - 30 м и диаметром 0,2 - 0,3 мм. Особенно рекомендуются готовые колонки с сорбентами класса полиорганосилоксанов зарубежного или отечественного производства (например, колонки из плавленого кварца с химически связанными НЖФ); их применение избавляет от необходимости проводить исключительно трудоемкую и скрупулезную работу подготовки хроматографической колонки достаточной эффективности.
Идентификацию ХОП и ПХБ, а также обработку хроматограмм ввиду значительной мультиплетности последних (например, стандартная смесь ПХБ хлофен А-50 разделяется на капиллярной колонке с DB-1 приблизительно на 50 компонентов (рис. 31)) целесообразно проводить с помощью ЭВМ, систем автоматического анализа либо, по меньшей мере, простейшего интегратора.

Рис. 31. Капиллярная хроматограмма смеси ПХБ хлофен A-50
(номера пиков ПХБ соответствуют классификации ИЮПАК).
По J.Duinker et al.,Analyt.Chem., 1988, v.60, p.478
Отбор проб воды, подготовку к анализу, подготовку посуды, экстракцию, концентрирование и очистку экстрактов, холостые определения, приготовление стандартных растворов хлорированных углеводородов и установление градуировочных характеристик метода, определение характеристик линейности диапазонов детектирования производят согласно гл. "Хлорированные углеводороды" с исключением любых связанных с дегидрохлорированием работ. Вследствие того, что применение настоящей методики позволяет вводить в детектор хроматографа существенно меньший объем аналитического раствора, чем в методике с применением набивной колонки, при экологических исследованиях приходится обрабатывать значительные массы морской воды (до 100 л и более). Извлекать анализируемые вещества из таких объемов воды экстракционным способом практически невозможно, поэтому рекомендуется использовать обработанные по специальным методикам сорбенты, такие как флоризил или ХАД-2.
Анализ хлорированных углеводородов на газовом хроматографе с капиллярной колонкой проводится по той же схеме, что и на приборе с набивной колонкой (за исключением дегидрохлорирования). При этом в случаях, когда необходимо количественно определять индивидуальные ПХБ, применяют предварительное фракционирование аналитических растворов методом адсорбционной колоночной хроматографии, для чего чаше всего используют активный силикагель (зернением 0,04 - 0,10 мм) и м-гексан и бензол в качестве подвижной фазы; в гексановую фракцию переходят в основном ПХБ, а в бензольную - ХОП группы дифенилэтана.
Измерение содержания хлорированных углеводородов на хроматографе с капиллярной колонкой рекомендуется проводить с программированием температуры в следующем оптимальном режиме: начальная температура термостата колонки 120°С (изотерма около 1 мин), далее нагрев со скоростью 5(...°)/мин до 250°С (изотерма около 10 мин); шкала электрометра - 10-12; расход газа-носителя (азота), соответствующий давлению 1,5 кг/см2.
Обработку результатов анализа вручную проводят согласно гл. "Хлорированные углеводороды", п. п. 8.1 - 8.2.
Затраты рабочего времени на анализ одной пробы морской воды по настоящей методике составляют около 20 чел.-ч.