СПРАВКА
Источник публикации
В данном виде документ опубликован не был.
Первоначальный текст документа опубликован в издании
М.: Федеральный центр гигиены и эпидемиологии Роспотребнадзора, 2010.
Информацию о публикации документов, создающих данную редакцию, см. в справке к этим документам.
Примечание к документу
Документ утратил силу в части гл. 1-3, 5-9, прил. 1 в связи с изданием Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
Документ введен в действие со 02.06.2010.
Взамен "Методических рекомендаций по определению вирулицидной активности препаратов" (от 06.09.1973 N 1119-73), "Инструкции по определению бактерицидных свойств новых дезинфицирующих средств" (от 06.05.1968 N 739-68), Методических указаний "Методы испытаний дезинфекционных средств для оценки их безопасности и эффективности", 1998 г.
Название документа
"Р 4.2.2643-10. 3.5. Дезинфектология. Методы лабораторных исследований и испытаний дезинфекционных средств для оценки их эффективности и безопасности. Руководство"
(утв. Роспотребнадзором 01.06.2010)
(ред. от 18.12.2020)
"Р 4.2.2643-10. 3.5. Дезинфектология. Методы лабораторных исследований и испытаний дезинфекционных средств для оценки их эффективности и безопасности. Руководство"
(утв. Роспотребнадзором 01.06.2010)
(ред. от 18.12.2020)
Руководитель Федеральной
службы по надзору в сфере
защиты прав потребителей
и благополучия человека,
Главный государственный
санитарный врач
Российской Федерации
Г.Г.ОНИЩЕНКО
1 июня 2010 года
3.5. ДЕЗИНФЕКТОЛОГИЯ
МЕТОДЫ ЛАБОРАТОРНЫХ ИССЛЕДОВАНИЙ И ИСПЫТАНИЙ
ДЕЗИНФЕКЦИОННЫХ СРЕДСТВ ДЛЯ ОЦЕНКИ
ИХ ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
РУКОВОДСТВО
Р 4.2.2643-10
Список изменяющих документов |
Дата введения
2 июня 2010 года
1. Разработаны Федеральным государственным учреждением науки "НИИ дезинфектологии" Роспотребнадзора и кафедрой дезинфектологии ММА им. И.М. Сеченова; Федеральной службой по надзору в сфере защиты прав потребителей и благополучия человека; ФГУН 48 НИИМ МО РФ - Центром военно-технических проблем биологической защиты; Научно-исследовательским центром бытовой химии; ООО "Суперсан-МК"; Институтом медицинской паразитологии тропической медицины им. Е.И. Марциновского ММА им. И.М. Сеченова; кафедрой тропической медицины и паразитарных болезней МПФ ППО ММА им. И.М. Сеченова; ГУ НИИ вирусологии им. Д.И. Ивановского РАМН; ГУ институтом полиомиелита и вирусных энцефалитов им. М.П. Чумакова РАМН.
2. Рекомендованы к утверждению Комиссией по санитарно-эпидемиологическому нормированию при Федеральной службе по надзору в сфере защиты прав потребителей и благополучия человека (протокол от 24 декабря 2009 г. N 4).
3. Утверждены Руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главным государственным санитарным врачом Российской Федерации Г.Г. Онищенко 1 июня 2010 г.
4. Введены в действие со 2 июня 2010 г.
5. Введены взамен "Методических рекомендаций по определению вирулицидной активности препаратов" (от 06.09.1973 N 1119-73), "Инструкции по определению бактерицидных свойств новых дезинфицирующих средств" (от 6 мая 1968 г. N 739-68), Методических указаний "Методы испытаний дезинфекционных средств для оценки их безопасности и эффективности", 1998 г.
Глава 1 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
Глава 2 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
Глава 3 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
дезинфекционных средств
Химические исследования являются важнейшей составляющей исследований и испытаний при создании, государственной регистрации, производстве, применении и сертификации дезинфекционных средств.
При создании дезинфекционных средств химические исследования включают:
- синтез действующего вещества;
- разработку рецептуры средства;
- разработку методов анализа действующих веществ и средств;
- установление срока годности действующих веществ и средств.
При государственной регистрации дезинфекционных средств проводят химико-аналитические исследования с целью:
- установления соответствия представляемых на регистрацию образцов дезинфекционных средств требованиям нормативной документации;
- определения количеств средства в воздухе и на обрабатываемых изделиях (санитарно-химические исследования) в случае использования в средствах летучих и плохо смываемых компонентов, представляющих потенциальную опасность при их использовании.
При производстве и применении дезинфекционных средств осуществляют контроль их качества согласно требованиям нормативной документации с использованием методов химико-аналитических исследований. При использовании дезинфицирующих средств в пищевой промышленности для обработки оборудования, кроме контроля качества самих дезинфицирующих средств, химическими методами контролируют также рабочие растворы средств и смывные воды с обрабатываемого оборудования.
При сертификационных испытаниях дезинфекционных средств химико-аналитические испытания являются основным способом подтверждения соответствия выпускаемого средства требованиям нормативной документации, т.е. соответствия выпускаемого средства зарегистрированному.
Дезинфекционные средства представляют собой составы в различных агрегатных состояниях: жидком, твердом и газообразном, а также в виде разных форм применения.
В состав дезинфекционных средств входят действующие вещества из разных классов химических соединений. Часто эти средства наряду с действующими веществами содержат функциональные добавки (моющие компоненты, синергисты, антикоррозионные добавки, растворители, регуляторы pH, красители, отдушки и пр.), придающие им дополнительные полезные свойства.
Поэтому для оценки качества дезинфекционных средств, помимо обязательного количественного определения действующих веществ, необходимо оценивать также косвенные интегральные физико-химические показатели качества, характеризующие многокомпонентные дезинфекционные средства в целом. Особенно характерно и обоснованно определение физико-химических показателей для дезинфицирующих средств. Эти показатели в совокупности в определенном приближении могут служить для дополнительной идентификации каждого конкретного дезинфицирующего средства.
Порядок определения показателей качества дезинфекционных средств следующий: сначала оценивают физико-химические показатели и только после полного соответствия этих показателей требованиям нормативной документации переходят к количественному анализу действующих веществ.
Такой порядок проведения химико-аналитических исследований дезинфекционных средств обусловлен тем, что при нем осуществляется переход от менее сложных и трудоемких в исполнении измерений, каковыми является определение физико-химических параметров, к более сложным процедурам количественного определения действующих веществ. Это позволяет обнаружить несоответствие средства требованиям нормативной документации уже на этапе измерений физико-химических параметров без проведения длительных исследований по количественному определению действующих веществ.
Контроль качества дезинфекционных средств химическими методами проводится с целью оценки соответствия средства требованиям нормативной документации и, следовательно, рецептуре средства. Вследствие этого, учитывая важную контролирующую функцию химических испытаний дезинфекционных средств, необходимо иметь методическую базу, позволяющую получать достоверные результаты при их аналитических исследованиях.
показателей дезинфекционных средств
К числу определяемых общих для всех средств показателей относятся внешний вид, включающий агрегатное состояние и цвет, а также запах. Эти показатели определяют органолептически.
Кроме того, для характеристики жидких дезинфекционных средств используют такие физико-химические показатели, как водородный показатель, называемый также показателем активности или показателем концентрации водородных ионов pH самого средства или его водных растворов; плотность при 20 °C; показатель преломления при 20 °C; относительную или удельную вязкость. Для порошкообразных индивидуальных веществ (субстанций) определяют температуру плавления и pH их водных растворов. Таблетированные формы дезинфицирующих средств испытывают на распадаемость и/или растворимость.
При этом, если указанные выше показатели качества (кроме водородного показателя) являются всего лишь интегральными характеристиками многокомпонентных рецептур, величина водородного показателя pH очень существенна для характеристики дезинфекционных средств, т.к. активность некоторых действующих веществ в значительной мере зависит от величины этого показателя. В то же самое время следует иметь в виду, что величиной pH не следует характеризовать средства с высоким содержанием органических растворителей, в частности, для рецептур с высоким содержанием спиртов величина pH является условной и ее не измеряют.
В связи с вышеизложенным перечень тестируемых физико-химических параметров определяется конкретно для каждого средства с учетом его агрегатного состояния и состава.
Органолептическую оценку, включающую визуальное определение внешнего вида и определение запаха обонянием, можно проводить в соответствии с ГОСТ 27025-86 [31]. В случае избрания других условий оценки внешнего вида и запаха, эти условия (посуда и освещение - при определении внешнего вида и особые условия определения запаха) должны быть указаны в нормативной документации.
Распадаемость и растворимость таблетированных форм дезинфицирующих средств определяют по времени распадения и растворения их в определенном объеме воды. Условия определения указанных показателей обычно устанавливают разработчики средств, при этом желательно, чтобы они были приближены к условиям применения этих средств.
Измерение физико-химических параметров - водородного показателя, плотности, показателя преломления, относительной или удельной вязкости, температуры плавления - проводят в соответствии с действующими ГОСТами на методы определений и Государственной фармакопеей СССР XI издания. В частности, водородный показатель определяют потенциометрически [33, 34, 46], плотность при 20 °C измеряют с помощью ареометра или пикнометра [36, 43], показатель преломления при 20 °C - рефрактометрически [37, 42], температуру плавления - термометрически [42, 44], вязкость - вискозиметрически [45].
действующих веществ в дезинфекционных средствах
Без контроля качества выпускаемой продукции - приемосдаточных и сертификационных испытаний на соответствие нормативной документации - не может быть использовано ни одно дезинфекционное средство. При этом обязательным условием оценки качества дезинфекционных средств является количественное определение действующих веществ, обусловливающих их активность и токсичность.
Вследствие этого нормативная документация на дезинфекционные средства (технические условия на отечественную и спецификации на зарубежную продукцию) должна в обязательном порядке включать нормы на содержание действующих веществ. Кроме того, представляемые зарубежными фирмами спецификации должны сопровождаться описаниями методик анализа действующих веществ.
Поскольку в качестве действующих веществ в дезинфекционных средствах применяются вещества из разных классов химических соединений, для количественного определения действующих веществ используются разнообразные методы химического анализа: гравиметрические, титриметрические, фотоколориметрические, спектрофотометрические, методы газовой - газоадсорбционной и газожидкостной хроматографии и высокоэффективной жидкостной хроматографии и др.
Методы и конкретные методики, которые могут быть использованы для количественного определения действующих веществ в средствах указанного назначения, многочисленны. Все они разные и потому не всегда поддаются унификации. В связи с этим ниже приводим систематизированный и обобщенный материал по методам анализа действующих веществ из различных классов химических соединений с описанием некоторых методик, наиболее применяемых для анализа дезинфекционных средств. Приведенные методики могут оказаться неприменимыми для многокомпонентных рецептур, содержащих неиндифферентные к конкретным методам ингредиенты. В таких случаях следует путем химических исследований выяснить причины искажающего действия компонентов на результаты анализов и устранить их в рамках существующих методик или разработать новые методы анализа.
Для анализа действующих веществ в дезинфекционных средствах могут быть использованы также другие, не приведенные ниже методики анализа, обеспечивающие достаточную точность, воспроизводимость и сходимость результатов.
В нижеследующих главах приведено описание методов анализа действующих веществ в дезинфицирующих, стерилизующих средствах, средствах для предстерилизационной очистки, дезинсекционных (инсектицидных, акарицидных, инсектоакарицидных и репеллентных) и дератизационных средствах.
Приводимые ниже методы - это наиболее часто применяемые для контроля качества дезинфекционных средств методы анализа, описанные в химико-аналитических разделах инструкций по их применению.
веществ в дезинфицирующих, стерилизующих средствах
и средствах для предстерилизационной очистки
В дезинфицирующих и стерилизующих средствах действующими веществами в основном являются:
- галоидактивные (хлор-, бром- и йодактивные соединения);
- кислородактивные соединения (перекись водорода, ее комплексы с солями, надуксусная кислота, озон);
- альдегиды (глутаровый альдегид, глиоксаль, формальдегид, янтарный альдегид, ортофталевый альдегид);
- четвертичные аммониевые соли (алкилдиметилбензиламмоний хлорид, дидецилдиметиламмоний хлорид и т.п. из ряда катионных ПАВ);
- производные гуанидина (соли полигексаметиленгуанидина, полигексаметиленбигуанидина и хлоргексидин биглюконат);
- третичный алкиламин - N,N-бис3-аминопропилдодециламин;
- низшие жирные спирты (этиловый, изопропиловый и н-пропиловый);
- фенольные соединения [2-бифенилол, 2-бензил-4-хлорфенол, 5-хлор-2-гидроксидифенилметан, 5-хлор-2-(2,4-дихлорфенокси)-фенол] и др.;
- кислоты органические и неорганические;
- щелочи;
- метасиликат натрия и др.
В состав средств для предстерилизационной очистки входят моющие компоненты, содержание которых в ряде случаев также контролируется. Это вещества из класса анионных, неионогенных и катионных ПАВ, щелочи, соли щелочных металлов (в частности, метасиликат натрия), ферменты и др.
Для количественного определения действующих веществ, входящих в состав дезинфицирующих, стерилизующих средств и средств для предстерилизационной очистки, в настоящее время наиболее часто применяют описанные ниже методы анализа.
(хлорактивных, бромактивных и йодактивных)
Анализ галоидактивных соединений (хлорактивных, бромактивных и йодактивных) основан на определении активных галоидов (активного хлора, активного брома и активного йода соответственно).
Из этого класса в качестве действующих веществ дезинфицирующих средств в основном используют хлорактивные и йодактивные соединения.
Определение хлорактивных соединений. Анализ хлорактивных соединений проводят с количественным определением активного хлора методом йодометрического титрования.
Сущность метода заключается в титровании серноватистокислым натрием (тиосульфатом натрия) свободного йода, выделяющегося при взаимодействии содержащих активный хлор соединений с йодистоводородной кислотой, образующейся из йодистого калия в кислой среде [35].
Выполнение анализа. К навеске средства или аликвоте раствора навески средства, содержащим около 35 мг активного хлора, прибавляют 10 см3 10%-го раствора йодистого калия, 10 см3 10%-го раствора серной кислоты, перемешивая после добавления каждого реактива, закрывают колбу пробкой и выдерживают в темном месте 3 мин.
Выделившийся йод титруют 0,1 н раствором тиосульфата натрия до светло-желтой окраски, прибавляют 1 - 2 см3 0,5%-го раствора крахмала и продолжают титровать до исчезновения синей окраски раствора.
Обработка результатов. Массовую долю активного хлора X в процентах вычисляют по формуле:
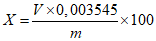 , где
, гдеV - объем раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,003545 - масса активного хлора, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н, г/см3;
m - масса анализируемой пробы, г.
Определение бромактивных соединений. Анализ бромактивных соединений проводят с количественным определением активного брома методом йодометрического титрования.
Выполнение анализа. По описанной для хлорактивных соединений методике анализируют навеску или аликвоту раствора навески, содержащие около 80 мг активного брома.
Обработка результатов. Массовую долю активного брома X в процентах вычисляют по формуле:
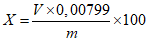 , где
, гдеV - объем раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,00799 - масса активного брома, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н, г/см3;
m - масса анализируемой пробы, г.
Определение йодактивных соединений. Для анализа йодактивных соединений также используют метод йодометрического титрования. Сущность метода заключается в титровании активного йода раствором тиосульфата натрия.
Выполнение анализа. Навеску средства, содержащую около 130 мг активного йода, вносят в титровальную колбу и доводят объем дистиллированной водой до 30 см3. Полученный раствор йодофора титруют 0,1 н раствором тиосульфата натрия в присутствии индикатора - 2 см3 (0,5%-го раствора крахмала), добавляемого при переходе окраски в светло-желтый цвет. Иногда титрование йодактивных соединений ведут в отсутствии крахмала до обесцвечивания раствора.
Обработка результатов. Массовую долю активного йода X в процентах вычисляют по формуле:
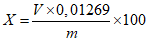 , где
, гдеV - объем раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,01269 - масса активного йода, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н, г/см3;
m - масса анализируемой пробы, г.
(перекиси водорода, ее комплексов с солями,
надуксусной кислоты и озона)
Определение активного кислорода. Часто перекисные соединения анализируются на содержание активного кислорода.
Для количественного определения активного кислорода используют метод йодометрического титрования [35].
Сущность метода заключается в титровании раствором тиосульфата натрия свободного йода, выделяющегося при взаимодействии содержащих активный кислород соединений с йодистоводородной кислотой, образующейся из йодистого калия в кислой среде.
Выполнение анализа. Навеску средства, содержащую около 8 мг активного кислорода, титруют в условиях, описанных в п. 2.1, для йодометрического титрования хлорактивных соединений [35].
Обработка результатов. Массовую долю активного кислорода X в процентах вычисляют по формуле:
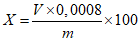 , где
, гдеV - объем раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,0008 - масса активного кислорода, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н, г/см3;
m - масса анализируемой пробы, г.
Методы определения перекиси водорода. Для анализа перекиси водорода могут быть использованы следующие объемные методы - перманганатометрическое, йодометрическое или периметрическое титрование. В литературе описан целый ряд других методов анализа перекиси водорода (фотоколориметрический, спектрофотометрический, полярографический и пр.).
Наиболее применяемым из указанных выше методов является метод перманганатометрического титрования, описанный в ГОСТе на перекись водорода [29].
Выполнение анализа. Навеску средства, содержащую около 25 мг перекиси водорода, помещают в коническую колбу вместимостью 250 см3, содержащую 25 см3 дистиллированной воды и 20 см3 раствора серной кислоты (разбавление 1:4 по объему), перемешивают и титруют 0,1 н раствором марганцовокислого калия до розовой окраски, не исчезающей в течение минуты.
Параллельно проводят контрольный опыт в тех же условиях и с тем же количеством реактивов, но без добавления перекиси водорода.
Обработка результатов. Массовую долю перекиси водорода X в процентах вычисляют по формуле:
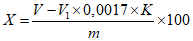 , где
, гдеV - объем раствора марганцовокислого калия концентрации с (1/5 KMnO4) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование анализируемой пробы, см3;
V1 - объем раствора марганцовокислого калия концентрации точно с (1/5 KMnO4) = 0,1 моль/дм3 (0,1 н), израсходованный на контрольное титрование, см3;
0,0017 - масса перекиси водорода, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (1/5 KMnO4) = 0,1 моль/дм3 0,1 н, г/см3;
K - поправочный коэффициент раствора марганцовокислого калия концентрации с (1/5 KMnO4) = 0,1 моль/дм3 0,1 н;
m - масса анализируемой пробы, г.
Методы определения надуксусной кислоты. Надуксусная (пероксоуксусная) кислота образуется при взаимодействии уксусной кислоты с перекисью водорода. Средства на ее основе получаются смешиванием этих компонентов, поэтому они практически всегда содержат перекись водорода.
Основными методами анализа систем перекись водорода - надуксусная кислота являются методы последовательного перманганатометрически-йодометрического и цериметрически-йодометрического титрования компонентов в одной и той же анализируемой пробе. Сначала перманганатометрическим или цериметрическим титрованием определяют перекись водорода, после чего проводят йодометрическое определение надуксусной кислоты. Для удаления кислорода, выделяющегося при перманганатометрическом и цериметрическом титровании перекиси водорода, к кислому титруемому раствору прибавляют карбонаты натрия. Выделяющийся при этом углекислый газ уносит с собой растворенный кислород.
Выполнение анализа. К навеске средства, содержащей перекись водорода и около 15 мг надуксусной кислоты, прибавляют 90 см3 10%-го раствора серной кислоты и проводят титрование перекиси водорода 0,1 н раствором марганцовокислого калия до светло-розового окрашивания, не исчезающего в течение минуты. К оттитрованной пробе прибавляют 1 г карбоната или бикарбоната натрия, колбу взбалтывают в течение 2 мин, после чего прибавляют 10 см3 10%-го раствора йодида калия и выдерживают 10 мин в темном месте.
Выделившийся йод титруют 0,1 н водным раствором тиосульфата натрия до светло-желтой окраски, прибавляют 1 см3 1%-го водного раствора крахмала и продолжают титровать до обесцвечивания раствора.
Обработка результатов. Массовую долю перекиси водорода в смеси ее с надуксусной кислотой вычисляют по формуле, приведенной выше в подразделе "Методы определения перекиси водорода".
Массовую долю надуксусной кислоты X в процентах вычисляют по формуле:
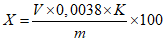 , где
, гдеV - объем раствора тиосульфата натрия концентрации с (Na2S2O3·5H2O) = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,0038 - масса надуксусной кислоты, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н, г/см3;
K - поправочный коэффициент раствора тиосульфата натрия концентрации с (Na2S2O3·5H2O) = 0,1 моль/дм3 0,1 н;
m - масса анализируемой пробы, г.
Методы определения озона. Наиболее доступны и применяемы в настоящее время методы количественного определения озона с помощью газоанализаторов.
Для анализа озона в жидких средах используют измеритель концентрации озона ИКО-3.
Принцип действия измерителя заключается в фотоколориметрическом определении озона по собственной полосе поглощения при длине волны 253,7 нм.
Диапазон измеряемой концентрации растворенного озона от 0,1 до 30 мг/л.
Выполнение измерений проводят согласно инструкции по применению прибора.
Для анализа озона в воздухе могут быть использованы газоанализаторы озона - измеритель концентрации озона в газе ИКО-1, оптический газоанализатор озона "Циклон-5.51" и хемилюминесцентный газоанализатор озона модели "3.02П-Р".
Принцип действия ИКО-1 и оптического газоанализатора заключается в фотоколориметрическом определении озона по собственной полосе поглощения при длине волны 253,7 нм.
Диапазон измеряемых концентраций озона прибором ИКО-1 - от 1 до 150 мг/л, газоанализатором "Циклон-5.51" - от 0 до 100 мг/м3.
Принцип действия газоанализатора озона модели "3.02П-Р" - в эффекте гетерогенной хемилюминесценции, возникающей в результате экзотермической реакции озона с окисляемыми химическими веществами композиции. Интенсивность свечения композиции пропорциональна концентрации озона в газовой смеси, измеряется и преобразуется в цифровой сигнал, отображаемый на мониторе анализатора.
Диапазон измеряемых концентраций - 0 - 500 мкг/м3.
Выполнение измерений указанными газоанализаторами озона проводят согласно руководствам по их эксплуатации.
глиоксаля, формальдегида, орто-фталевого альдегида и др.)
Альдегиды, входящие в состав дезинфицирующих средств, анализируют в основном следующими методами: объемными (титриметрическими) - бисульфитным (2 варианта), сульфитным и с использованием гидроксиламина и гидроксиламина солянокислого. В последние годы для их анализа стали использовать методы ГЖХ и ВЭЖХ, которые приводятся в инструкциях по применению дезинфицирующих средств.
Сущность бисульфитного метода заключается в образовании бисульфитного производного при взаимодействии альдегидов с бисульфитом натрия с последующим определением альдегидов методом йодометрического титрования. В первом классическом варианте метода используется обратное титрование прибавляемого избытка йода раствором тиосульфата натрия. Второй вариант предусматривает прямое титрование раствором йода.
В основе сульфитного метода - образование при взаимодействии альдегидов с сульфитом натрия гидроокиси натрия, титруемой растворами серной или соляной кислот.
При применении в анализе свободного гидроксиламина в спиртовой среде происходит оксимирование, и содержание альдегида определяется разностью объемов соляной кислоты, израсходованных на титрование в контрольном опыте и при титровании анализируемой пробы.
Сущность метода с использованием гидроксиламина солянокислого заключается в том, что при взаимодействии альдегидов с ним образуются альдоксимы с выделением эквивалентного количества соляной кислоты, которая титруется раствором гидроокиси натрия, либо потенциометрически до pH в интервале от 3 до 4, либо с индикатором бромфеноловым синим.
Ниже приводится в обобщенном виде наиболее часто применяемый метод количественного определения альдегидов с использованием гидроксиламина солянокислого на примере глутарового альдегида.
Выполнение анализа. Навеску или аликвоту навески средства, содержащие около 250 мг глутарового альдегида, вносят в коническую колбу с притертой пробкой, прибавляют 10 см3 дистиллированной воды и 0,1 см3 0,1%-го водного раствора бромфенолового синего.
В случае кислой реакции полученного раствора (раствор в присутствии индикатора окрашивается в желтый цвет) прибавляют 0,5 н раствор гидроокиси натрия до появления синего окрашивания. В случае щелочной реакции раствора (раствор окрашивается в синий цвет) прибавляют 0,5 н раствор соляной кислоты до светло-желтого окрашивания и затем 0,5 н раствор гидроокиси натрия до появления синего окрашивания. Затем вносят 25 см3 7%-го водного раствора гидроксиламина солянокислого, закрывают колбу пробкой, оставляют на 20 - 30 мин при комнатной температуре, после чего образовавшийся раствор желтого цвета титруют 0,5 н раствором гидроокиси натрия до появления синего окрашивания.
Обработка результатов. Массовую долю глутарового альдегида X в процентах вычисляют по формуле:
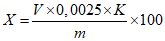 , где
, гдеV - объем раствора гидроокиси натрия концентрации с NaOH = 0,5 моль/дм3 (0,5 н), израсходованный на титрование, см3;
0,0025 - масса глутарового альдегида, соответствующая 1 см3 раствора тиосульфата натрия концентрации точно с NaOH = 0,5 моль/дм3 (0,5 н), г/см3;
K - поправочный коэффициент раствора гидроокиси натрия концентрации с NaOH = 0,5 моль/дм3 (0,5 н);
m - масса анализируемой пробы, г.
Анализ четвертичных аммониевых солей (ЧАС) из ряда катионных ПАВ в дезинфицирующих средствах проводят с использованием методов двухфазного и аргентометрического титрования, фотоколориметрического, спектрофотометрического и других методов.
В большинстве случаев для анализа ЧАС применяют методики анализа, основанные на методе двухфазного титрования.
Существует много вариантов метода двухфазного титрования ЧАС, отличающихся по величине pH титруемого раствора и по используемым индикаторам. Для анализа дезинфицирующих средств применяют щелочное и кислотное титрования с использованием индикаторов бромфенолового синего, метиленового голубого, смешанных индикаторов метиленового голубого и эозина Н, а также димидиум бромида и дисульфина голубого.
В основе метода количественного определения ЧАС двухфазным титрованием лежит взаимодействие катионоактивных ЧАС с анионоактивным додецилсульфатом натрия.
Указанные варианты двухфазного титрования позволяют количественно определять ЧАС с большой степенью точности. Они описаны в технических условиях и инструкциях по применению дезинфицирующих средств, содержащих ЧАС.
Ниже приведена методика анализа алкилдиметилбензиламмоний хлорида методом двухфазного титрования в щелочной среде с использованием индикатора бромфенолового синего.
Приготовление щелочного буферного раствора с pH 11. 50 г сульфата натрия и 3,5 г карбоната натрия растворяют в 500 см3 дистиллированной воды.
Выполнение анализа. В цилиндр или коническую колбу вносят навеску или аликвоту раствора навески средства, содержащие около 15 мг алкилдиметилбензиламмоний хлорида, 30 см3 буферного раствора с pH 11, 20 см3 хлороформа и 8 - 12 капель 0,1%-го водного раствора индикатора бромфенолового синего. Титруют 0,004 н раствором додецилсульфата натрия с интенсивным встряхиванием в закрытой емкости до появления отчетливого фиолетового окрашивания в верхней водной фазе.
Обработка результатов. Массовую долю алкилдиметилбензиламмоний хлорида X в процентах вычисляют по формуле:
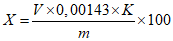 , где
, гдеV - объем раствора додецилсульфата натрия концентрации с (C12H25OSO3Na) = 0,004 моль/дм3 (0,004 н), израсходованный на титрование, см3;
0,0038 - масса алкилдиметилбензиламмоний хлорида, соответствующая 1 см3 раствора додецилсульфата натрия концентрации точно с (C12H25OSO3Na) = 0,004 моль/дм3 (0,004 н), г/см3, при средней молекулярной массе ЧАС 357, г/см3;
K - поправочный коэффициент раствора додецилсульфата натрия концентрации с (C12H25OSO3Na) = 0,1 моль/дм3 (0,1 н);
m - масса анализируемой пробы, г.
Данный метод не избирателен в присутствии ПГМГ.
(солей полигексаметиленгуанидина гидрохлорида
и хлоргексидина биглюконата)
Методы определения полигексаметиленгуанидин гидрохлорида и полигексаметиленгуанидин фосфата. В настоящее время для анализа солей полигексаметиленгуанидина в дезинфицирующих средствах часто используют фотоколориметрический метод с использованием красителя эозина Н. Для анализа солей полигексаметиленгуанидина в составе дезинфицирующих средств разработан также метод двухфазного титрования.
Разные модификации этих методов описаны в инструкциях по применению средств, содержащих соли полигексаметиленгуанидина.
Ниже приведен один из вариантов фотоколориметрической методики анализа полигексаметиленгуанидин гидрохлорида ПГМГ.
Данная методика может быть использована только при отсутствии в составе средства других азотсодержащих органических соединений (ЧАС, аминов и др.).
Сущность метода заключается в фотометрическом определении оптической плотности ионных ассоциатов, образуемых в растворах при взаимодействии ПГМГ с эозином Н.
Приготовление буферного раствора. Готовят два раствора:
Раствор 1 - 0,1 н водный раствор соляной кислоты с использованием фиксанала.
Раствор 2 - 0,75 г глицина и 0,59 г хлористого натрия растворяют в мерной колбе вместимостью 100 см3 с доведением объема дистиллированной водой до метки.
Для приготовления буферного раствора в мерную колбу вместимостью 100 см3 вносят 92,5 см3 раствора 2 и объем жидкости доводят до метки раствором 1. Значение pH буферного раствора должно быть 3,5 +/- 0,1, что необходимо контролировать с помощью pH-метра.
Построение градуировочного графика. График строят в интервале концентраций от 0,4 до 1,6 мкг/см3. Для этого в мерные колбы вместимостью 25 см3 вносят 1, 2, 3 и 4 см3 раствора, содержащего 10 мкг/см3 стандартного образца ПГМГ, и доводят объемы во всех колбах до 10 см3 прибавлением соответственно 9, 8, 7 и 6 см3 дистиллированной воды. Затем прибавляют 1 см3 0,05%-го водного раствора эозина Н, 10 см3 глицинового буферного раствора с pH 3,5 и доводят объем дистиллированной водой до метки. После перемешивания растворы фотометрируют относительно образца сравнения при длине волны 540 нм в кюветах с толщиной поглощающего слоя 50 мм.
Образец сравнения готовят внесением в мерную колбу вместимостью 25 см3 10 см3 дистиллированной воды, 1 см3 0,05%-го раствора эозина Н, 10 см3 буферного раствора с доведением объема дистиллированной водой до метки.
Выполнение анализа. Навеску средства, содержащую 20 - 30 мг ПГМГ, разводят или растворяют в мерной колбе вместимостью 100 см3 с доведением объема дистиллированной водой до метки.
1 см3 приготовленного раствора разводят дистиллированной водой в мерной колбе вместимостью 100 см3 до метки. Берут 10 см3 раствора второго разведения, вносят в мерную колбу вместимостью 25 см3 и далее прибавляют раствор эозина Н, буферный раствор и проводят определение оптической плотности в указанных выше для построения градуировочного графика условиях.
Обработка результатов. Массовую долю ПГМГ X в процентах вычисляют по формуле:
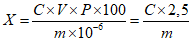 , где
, гдеC/10-6 - концентрация ПГМГ, обнаруженная по градуировочному графику в анализируемой пробе, г/см3;
V - объем раствора анализируемой пробы, равный 100 см3;
P - кратность разведения раствора анализируемой пробы, равная 250 см3;
m - масса анализируемой пробы, г.
Методы определения полигексаметиленбигуанидина гидрохлорида. Полигексаметиленбигуанидин гидрохлорид, называемый бигуанидом, олигомером бигуанида или полигексаметиленбигуанидом, может быть количественно определен фотоколориметрически по методике, описанной выше для солей полигексаметиленгуанидина.
Кроме того, для его анализа может быть использован спектрофотометрический метод с проведением измерений при длине волны 233 - 237 нм.
Методы определения хлоргексидина биглюконата. Количественное определение хлоргексидина биглюконата в дезинфицирующих средствах проводят с использованием следующих методов: метода безводного титрования хлорной кислотой, фотоколориметрического метода, основанного на окрашивании хлоргексидина биглюконата гипобромитом натрия, спектрофотометрического метода и метода ВЭЖХ. Описание последних двух методов дается в ряде инструкций по применению дезинфицирующих средств.
Приводим описание спектрофотометрического метода анализа хлоргексидина биглюконата.
Выполнение анализа. Навеску средства, содержащую около 1 мг хлоргексидина биглюконата, помещают в мерную колбу вместимостью 100 см3, объем доводят дистиллированной водой до метки и перемешивают.
Измеряют оптическую плотность полученного раствора на спектрофотометре при длине волны 253 нм в кювете с толщиной поглощающего слоя 1 см.
В качестве раствора сравнения используют дистиллированную воду.
Обработка результатов. Массовую долю хлоргексидина биглюконата X в процентах вычисляют по формуле:
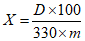 , где
, гдеD - оптическая плотность испытуемого раствора;
330 - удельный показатель поглощения хлоргексидин биглюконата;
m - масса анализируемой пробы, г.
N,N-бис(3-аминопропил)-додециламина
Для количественного определения указанного третичного алкиламина в дезинфицирующих средствах используют метод кислотно-основного титрования, в т.ч. потенциометрическое титрование в спиртовой среде. Кроме того, могут быть использованы метод неводного титрования хлорной кислотой в присутствии индикатора кристаллического фиолетового и метод ГЖХ.
В случае отсутствия в средствах кислых и щелочных компонентов анализ третичного амина проводят описанным ниже методом кислотно-основного титрования в водной среде.
Сущность метода заключается в титровании амина раствором соляной кислоты, с которой амин реагирует с образованием соответствующей соли.
Выполнение анализа. В титровальную колбу вносят навеску средства, содержащую около 100 мг третичного амина, доводят объем дистиллированной водой до 30 см3, вносят 1,0 см3 0,1%-го водного раствора индикатора бромтимолового синего и титруют 0,1 н раствором соляной кислоты до перехода синей окраски в желтую.
Обработка результатов. Массовую долю N,N-бис(3-аминопропил)-додециламина (X) в процентах вычисляют по формуле:
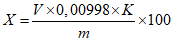 , где
, гдеV - объем раствора соляной кислоты концентрации с HCl = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,00998 - масса N,N-бис3-аминопропилдодециламина, соответствующая 1 см3 раствора соляной кислоты концентрации точно с HCl = 0,1 моль/дм3 (0,1 н), г/см3;
K - поправочный коэффициент раствора соляной кислоты с HCl = 0,1 моль/дм3 (0,1 н);
m - масса анализируемой пробы, г.
этилового, изопропилового и н-пропилового
Для анализа низших жирных спиртов каждого в отдельности и их смесей используют газохроматографические методы, в основе которых лежит метод, описанный для парфюмерно-косметических изделий [32].
Количественная оценка спиртов - методом "внутреннего эталона", в качестве которого для этилового и изопропилового спиртов может быть использован н-пропиловый спирт, для н-пропилового спирта - изопропиловый или этиловый спирт.
Средства измерений. Хроматограф лабораторный газовый с пламенно-ионизационным детектором; колонка из нержавеющей стали длиной 200 см и внутренним диаметром 0,3 см, насадка - полисорб-1 с частицами размером 0,1 - 0,3 мм.
Условия хроматографирования:
Газ-носитель | азот |
Температура термостата | 30 °C |
Температура испарителя | 200 °C |
Температура детектора | 200 °C |
Предел измерения по току, А | 5 x 10-8 |
Объемный расход газа-носителя, см3/мин | 40 |
Объемный расход водорода, см3/мин | 60 |
Объемный расход воздуха, см3/мин | 300 |
Скорость движения ленты самописца, мм/ч | 240 |
Объем пробы | (0,6 - 1,0) см-3 |
Определение калибровочного коэффициента. Готовят две искусственные смеси взвешиванием равных количеств анализируемого спирта и внутреннего эталона. Каждую из них хроматографируют 10 раз.
Относительный калибровочный коэффициент рассчитывают по формуле:
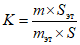 , где
, гдеm и mэт - массы определяемого спирта и внутреннего эталона с учетом чистоты, г;
S и Sэт - площади пиков определяемого спирта и внутреннего эталона, мм2.
Выполнение анализа. К анализируемому образцу прибавляют внутренний эталон в количестве, примерно равном определяемому компоненту. Готовят две пробы анализируемого образца и каждую из них хроматографируют 3 раза.
Обработка результатов. Массовую долю спирта X в процентах вычисляют по формуле:
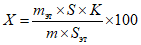 , где
, гдеm - масса анализируемого образца, г;
mэт - масса внутреннего эталона с учетом чистоты, г;
S - площадь пика определяемого спирта, мм2;
Sэт - площадь пика внутреннего эталона, мм2;
K - относительный калибровочный коэффициент.
Для количественной оценки спиртов применяют также метод абсолютной градуировки.
[2-бифенилола, 2-бензил-4-хлорфенола,
5-хлор-2-гидроксидифенил-метана,
5-хлор-2-2,4-дихлорфеноксифенола] и др.
Для определения о-фенилфенола, о-бензил-р-хлорфенола, 2-феноксиэтанола, 2-феноксипропанола, 5-хлор-2(2,4-дихлорфенокси)фенола триклозана в дезинфицирующих средствах могут быть применены спектрофотометрические и хроматографические методы в зависимости от состава и концентраций ДВ, а также вспомогательных веществ рецептурного состава средства.
Спектрофотометрические методы определения фенольных соединений проводят с прямым определением вещества по собственному поглощению или в виде окрашенных комплексов.
Спектрофотометрический метод определения фенольных соединений по собственному поглощению основан на измерении оптической плотности раствора средства при длине волны 260 - 290 нм относительно растворителя с применением градуировочного графика или молярных и удельных коэффициентов погашения.
Метод может быть применен для серийного анализа в условиях производства.
Метод не избирателен в присутствии компонентов рецептурного состава средства, поглощающих в указанной области длин волн, несколько фенольных соединений при совместном присутствии определяются суммарно.
Подготовка пробы к анализу зависит от рецептурного состава средства.
Так при определении о-фенилфенола по собственному поглощению в дезинфицирующем средстве на основе водного раствора пропилового и этилового спиртов пробу готовят разбавлением средства в этиловом спирте [21] и измеряют оптическую плотность при длине волны 286 нм, раствор сравнения - этиловый спирт.
При определении триклозана по собственному поглощению в антибактериальном туалетном мыле подготовка пробы включает экстракционное извлечение триклозана гексаном [22] для устранения мешающего влияния компонентов рецептурного состава, измерение оптической плотности проводят при длине волны 286 нм, раствор сравнения - гексан.
В случае совместного присутствия в дезинфицирующем средстве наряду с фенольными соединениями другого ДВ, характеризующегося значительно меньшими, чем у фенольных ДВ коэффициентами погашения, например 2-феноксипропанола и катионных ПАВ с ароматическими циклами, применяют большое разбавление исследуемого раствора (от 1:100 до 1:5000) [1].
Спектрофотометрическое определение триклозана в антибактериальном мыле может быть проведено по поглощению окрашенных продуктов его взаимодействия с 4-аминоантипирином в присутствии гексацианоферрата калия при 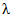 = 490 нм [21]. При этом градуировочные смеси для построения градуировочного графика необходимо готовить путем добавления известных количеств триклозана в водный раствор такого мыла, не содержащего триклозан.
= 490 нм [21]. При этом градуировочные смеси для построения градуировочного графика необходимо готовить путем добавления известных количеств триклозана в водный раствор такого мыла, не содержащего триклозан.
При использовании градуировочного графика в координатах "оптическая плотность - содержание вещества в аликвотной части раствора, мкг" массовую долю определяемого ДВ (X, %) вычисляют по формуле общего вида:
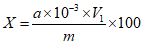 , где
, гдеa - содержание ДВ в аликвотной части раствора, найденное по градуировочному графику, мг;
10-3 - фактор пересчета из мг в г;
V1 - объем раствора средства или экстракта, мл;
m - масса средства, взятая на анализ, г.
При использовании значений молярных (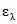 ) или удельных (
) или удельных (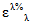 ) коэффициентов погашения массовую долю определяемого ДВ X, % вычисляют по формуле общего вида:
) коэффициентов погашения массовую долю определяемого ДВ X, % вычисляют по формуле общего вида:
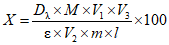 , где
, гдеM - молярная масса определяемого вещества ДВ, г/моль;
V1 - объем раствора или экстракта, мл;
V3 - объем фотометрируемого раствора, л;
m - масса средства, взятая для анализа, г;
V2 - объем отобранной аликвотной части раствора или экстракта, л;
l - толщина поглощающего слоя кюветы, см.
В [19, 21] приведены оценочные значения молярных коэффициентов погашения некоторых фенольных соединений при длине волны измерения 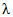 ,
, 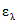 , л/моль·см: о-фенилфенол
, л/моль·см: о-фенилфенол 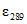 = 11000; о-бензил-р-хлорфенол
= 11000; о-бензил-р-хлорфенол 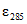 = 5300; 5-хлор-2(2,4-дихлорфенокси)-фенол
= 5300; 5-хлор-2(2,4-дихлорфенокси)-фенол 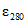 = 4500; 2-феноксипропанол
= 4500; 2-феноксипропанол 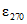 = 2000.
= 2000.
Приборы, реактивы и растворы. Для выполнения анализа применяют спектрофотометр СФ-26, СФ-46; электрофотоколориметр типа КФК-2, стандартную стеклянную мерную посуду, весы аналитические; примененный растворитель, реактивы для получения окрашенных соединений.
Хроматографические методы определения фенольных соединений. Проблемы анализа фенольных соединений, в т.ч. раздельного определения нескольких фенольных соединений при совместном присутствии, успешно решаются методами ГЖХ с применением пламенно-ионизационного детектирования и ВЭЖХ с применением спектрофотометрического детектирования. Современные хроматографы, снабженные программой управления, сбора и обработки хроматографических данных на базе персонального компьютера, позволяют достаточно быстро подбирать оптимальные условия анализа, гибко изменять программу температурных условий хроматографирования или создавать градиентный режим элюирования, проводить обработку хроматограмм в расширенном диапазоне концентраций определяемых веществ.
При анализе фенольных соединений методом ВЭЖХ применяют хроматографические колонки для обращенно-фазной ВЭЖХ типа Zorbax ODS, Zorbax Eclipse - XDB C8 3,5 мкм, Nucleosil 100 10 C18, LiChrospher 100 RP 18 5 мкм, Intersil ODS-2 и др. В качестве подвижной фазы наиболее часто используют: для изократического хроматографирования - растворы в соотношении по объему метанол:вода (90:10), ацетонитрил:вода (35:65), ацетонитрил:0,2%-й водный раствор ортофосфорной кислоты (68:32); для градиентного хроматографирования элюенты - метанол и 0,2%-й водный раствор хлорной кислоты, ацетонитрил и 1%-й водный раствор уксусной кислоты, ацетонитрил и 0,2%-й водный раствор ортофосфорной кислоты и др.
Для разделения фенольных соединений в ГЖХ применяют насадочные стандартные колонки длиной 1 м, внутренним диаметром 0,2 - 0,4 мм, в качестве неподвижной фазы используют СКТФ и силиконы SE-30 и XE-60, при содержании 3 - 5% к весу твердого носителя - силанизированного хроматона N-AW-DMCS и инертона AW-DMCS, зернением 0,20 - 0,25 мм, для анализа 2-феноксипропанола и 2-феноксиэтанола известно использование в качестве сорбента также карбосфера, пропитанного 0,1% AT-100 и др.
Разделение смеси действующих веществ 4-хлор-3-метилфенола и о-фенилфенола и глутарового альдегида получено на капиллярной колонке (50 м x 0,32 мм) с неподвижной фазой CP Sil 5 CB, толщиной слоя 1,2 мкм в режиме программирования температуры от 100 до 250 °C при скорости нагрева 10 °C/мин; время удерживания глутарового альдегида составило 6,4 мин, 4-хлор-3-метилфенола 13,4 мин и о-фенилфенола 16,8 мин.
Выбор режима хроматографирования - изотермического или программирования температуры - зависит от содержания, состава ДВ, а также вспомогательных компонентов рецептурного состава дезинфицирующего средства.
Количественную оценку определяемого вещества проводят с использованием абсолютной градуировки, которая целесообразна для выборочного единичного анализа, или с применением внутреннего эталона, рекомендуемого при выполнении массовых рутинных анализов, например в условиях производственного контроля.
Газохроматографическое определение 2-феноксиэтанола в кожном антисептике в виде геля [23] проводят с применением пламенно-ионизационного детектирования, изотермического хроматографирования раствора пробы после разрушения структуры геля и использованием абсолютной градуировки или внутреннего эталона, в качестве которого применяют 1-тетрадеканол (спирт тетрадециловый).
Приборы, реактивы и растворы. Газовый хроматограф, снабженный пламенно-ионизационным детектором, стандартной колонкой (1 м x 3 мм), заполненной силанизированным хроматоном N-AW-DMCS с 5% неподвижной фазы XE-60; стандартная стеклянная мерная посуда; микрошприц типа МШ вместимостью 1 мкл; спирт этиловый ректификованный с объемной долей не менее 96%; аналитический стандарт 2-феноксиэтанола; 1-тетрадеканол; газ-носитель - азот, водород из баллона, воздух от компрессора.
Выполнение анализа. К 50 г анализируемого средства добавляют около 0,25 г хлористого натрия, смесь перемешивают и образовавшийся раствор отфильтровывают через бумажный фильтр. Около 20 г фильтрата взвешивают с точностью до четвертого десятичного знака, добавляют 5 мл раствора тетрадецилового спирта и после перемешивания вводят в хроматограф.
Градуировочные смеси и анализируемую пробу хроматографируют при следующих условиях: расход газов - азот 45 мл/мин, водород 30 мл/мин, воздух 300 мл/мин; температура колонки (140 +/- 3) °C, испарителя 250 °C; объем вводимой дозы 1 мкл.
Приготовление градуированных смесей с внутренним эталоном. Для определения градуировочного коэффициента 2-феноксиэтанола по веществу - внутреннему эталону готовят три градуировочные смеси и раствор вещества-эталона: 0,14, 0,17 и 0,20 г 2-феноксиэтанола растворяют в 20 г этилового спирта; 4 г тетрадецилового спирта растворяют в 100 мл этилового спирта в мерной колбе. Результаты взвешиваний записывают с точностью до четвертого десятичного знака. К каждой градуировочной смеси 2-феноксиэтанола добавляют по 5 мл раствора тетрадецилового спирта, тщательно перемешивают и вводят в хроматограф не менее трех раз. Из каждой хроматограммы определяют площадь хроматографических пиков 2-феноксиэтанола, тетрадецилового спирта в градуировочной смеси и вычисляют градуировочный коэффициент K по формуле:
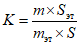 , где
, гдеm - масса 2-феноксиэтанола в градуировочной смеси, г;
mэт - масса вещества-эталона в 5 мл раствора тетрадецилового спирта, г;
S и Sэт - площади пиков 2-феноксиэтанола и вещества-эталона.
За градуировочный коэффициент (K) принимают среднее арифметическое результатов всех определений, расхождения которых от средней величины не превышают 20%.
Обработка результатов. Массовую долю 2-феноксиэтанола в средстве X, % вычисляют по формуле:
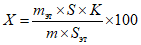 , где
, гдеK - градуировочный коэффициент 2-феноксиэтанола;
S и Sэт - площади пиков 2-феноксиэтанола и вещества-эталона в анализируемой пробе;
m и mэт - масса средства, взятая на анализ, и масса вещества-эталона в 5 мл раствора тетрадецилового спирта, г.
Газохроматографическое определение о-фенилфенола и о-бензил-р-хлорфенола, 3-метил-4-хлорфенола с применением пламенно-ионизационного детектирования, хроматографирования в режиме программирования температуры и использованием абсолютной градуировки.
Приборы, реактивы и растворы. Газовый хроматограф снабженный пламенно-ионизационным детектором, стандартной колонкой (1 м x 3 мм), заполненной силанизированным хроматоном N-AW-DMCS с 5% неподвижной фазы XE-60; стандартная стеклянная мерная посуда; микрошприц типа МШ вместимостью 1 мкл; ацетон; аналитические стандарты о-фенилфенола и о-бензил-р-хлорфенола; азот из баллона, водород из баллона, воздух от компрессора.
Выполнение анализа [24]. Около 3,3 г средства, взвешенного с точностью до четвертого десятичного знака, растворяют в ацетоне в мерной колбе вместимостью 25 мл и после перемешивания вводят в хроматограф.
Градуировочные смеси и анализируемую пробу хроматографируют при следующих условиях. Расход газов: азот 30 - 40 мл/мин, водород 25 - 30 мл/мин, воздух 250 - 300 мл/мин; температура испарителя и детектора 250 °C; температура колонки начальная 70 °C, нагрев со скоростью 15 °C/мин до 190 °C; объем вводимой дозы 1 мкл. Порядок выхода определяемых веществ: о-бензил-р-хлорфенол, о-фенилфенол.
Градуировочные смеси готовят с массовой концентрацией по 4 - 4,5 мг/мл о-фенилфенола и о-бензил-р-хлорфенола в ацетоне.
Обработка результатов. Массовую долю определяемого вещества в средстве X, % вычисляют по формуле:
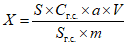 , где
, гдеS и Sг.с. - площади хроматографических пиков определяемого вещества в анализируемой пробе и градуировочной смеси;
Cг.с. - массовая концентрация аналитического стандарта определяемого вещества в градуировочной смеси, мг/мл;
a - массовая доля основного вещества в аналитическом стандарте, %;
V - объем раствора пробы, мл;
m - масса средства, взятая на анализ, мг.
Количественное определение кислот и щелочей проводят методом кислотно-основного титрования с использованием индикаторов кислотно-основного титрования, из которых наиболее часто применяются метиловый оранжевый, фенолфталеин, метиловый красный, бромфеноловый синий и др.
Приводится обобщенный метод количественного определения кислот на примере серной кислоты.
Сущность метода - в проведении реакции нейтрализации серной кислоты щелочью путем титрования раствором гидроокиси натрия в присутствии индикаторов кислотно-основного титрования.
Выполнение анализа. Навеску средства, содержащую около 50 мг серной кислоты, помещают в коническую колбу, прибавляют дистиллированную воду до 30 см3, необходимый объем раствора индикатора и титруют 0,1 н раствором гидроокиси натрия до соответствующего перехода окраски индикатора.
Обработка результатов. Массовую долю серной кислоты X в процентах вычисляют по формуле:
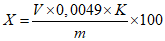 , где
, гдеV - объем раствора гидроксида натрия концентрации с NaOH = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,0049 - масса серной кислоты, соответствующая 1 см3 раствора гидроокиси натрия концентрации точно с NaOH = 0,1 моль/дм3 0,1 н, г/см3;
K - поправочный коэффициент раствора гидроокиси натрия концентрации с NaOH = 0,1 моль/дм3 (0,1 н);
m - масса анализируемой пробы, г.
Приводится обобщенный метод количественного определения щелочей на примере гидроокиси натрия.
Сущность метода в проведении реакции нейтрализации гидроокиси натрия кислотой путем титрования 0,1 н водным раствором соляной кислоты в присутствии индикаторов кислотно-основного титрования.
Выполнение анализа. Навеску средства, содержащую около 40 мг щелочи, помещают в коническую колбу, прибавляют дистиллированную воду до 30 см3, необходимый объем раствора индикатора и титруют 0,1 н раствором соляной кислоты до соответствующего перехода окраски индикатора.
Обработка результатов. Массовую долю гидроокиси натрия X в процентах вычисляют по формуле:
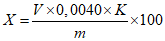 , где
, гдеV - объем раствора соляной кислоты концентрации с HCl = 0,1 моль/дм3 (0,1 н), израсходованный на титрование, см3;
0,0040 - масса гидроокиси натрия, соответствующая 1 см3 раствора соляной кислоты концентрации точно с HCl = 0,1 моль/дм3 0,1 н, г/см3;
K - поправочный коэффициент раствора соляной кислоты концентрации с HCl = 0,1 моль/дм3 0,1 н;
m - масса анализируемой пробы, г.
Количественное определение анионных ПАВ проводят с использованием метода двухфазного титрования в присутствии смешанного индикатора метиленового голубого и эозина БА или эозина Н, а также фотоколориметрическим методом, основанным на образовании окрашенного комплекса с азуром 1 с фотоколориметрическим измерением оптической плотности его хлороформного экстракта, согласно ГОСТам на указанные методы [39].
Для анализа неионогенных ПАВ (оксиэтилированных высших спиртов и алкилфенолов) может быть использован метод, сущность которого в осаждении неионогенного ПАВ избытком фосфорномолибденовой кислоты и последующим потенциометрическим титрованием ее избытка раствором диантипирилметана с катодно-поляризованным платиновым электродом [40].
Анализ метасиликата натрия проводят фотоколориметрическим методом, основанным на образовании окрашенного комплекса с молибденовокислым аммонием и гравиметрическим методом, сущность которого в осаждении кремниевой кислоты с определением ее массы. Описание этих методов приводится в ГОСТах на методы анализа силиката натрия [25].
Дезинсекционные средства включают инсектицидные, акарицидные, инсектоакарицидные и репеллентные средства.
В инсектицидных средствах в качестве действующих веществ ДВ используют пиретроиды, фосфорорганические соединения ФОС, карбаматы, регуляторы развития насекомых и некоторые вещества из других классов химических соединений в основном сложного химического строения, например фипронил, тиаметоксам, имидаклоприд, гидраметилнон.
В акарицидных и инсектоакарицидных средствах в качестве ДВ применяют пиретроиды и фосфорорганические соединения.
В репеллентных средствах действующими веществами в основном являются диметилфталат, N,N-диэтил-м-толуамид ДЭТА и этил-3-N-бутил-ацетамино-пропионат ИР3535.
Формами применения дезинсекционных средств являются: жидкие (лосьоны, эмульсии, шампуни, концентраты эмульсий, растворы в беспропеллентных аэрозольных упаковках БАУ); твердые (смачивающиеся порошки, дусты, карандаши, таблетки, шашки); гели; кремы; аэрозольные баллоны, электрофумигирующие средства (пластины, спирали, жидкости); свечи; а также бумага, ткани, салфетки, пропитанные инсектицидными составами, и др.
К основным показателям качества дезинсекционных средств, контролируемым при регистрации и сертификации, относятся: внешний вид, определяемый визуально, запах, оцениваемый органолептически, и массовая доля ДВ; к дополнительным при характеристике водно-спиртовых лосьонов и шампуней - показатель активности водородных ионов pH, определяемый потенциометрически по ГОСТ Р 50550-93 [33].
Для идентификации и количественного определения ДВ в дезинсекционных средствах используются методы газожидкостной хроматографии ГЖХ, высокоэффективной жидкостной хроматографии ВЭЖХ и спектрофотометрический.
Универсальным и наиболее доступным методом анализа ДВ в дезинсекционных средствах является метод ГЖХ, поскольку его можно использовать для количественного определения практически всех применяемых ДВ (за исключением имидаклоприда, гидраметилнона, определяемых методом ВЭЖХ, и азаметифоса, тиаметоксама, определяемых спектрофотометрическим методом).
Преимуществом метода ГЖХ является возможность эффективно разделять смеси ДВ в одном средстве, что затруднено при анализе спектрофотометрическим методом. Метод ГЖХ обладает высокой чувствительностью (0,01 - 0,05 мг/см3), что позволяет определять самые низкие концентрации ДВ в различных формах применения.
Погрешность метода определения ДВ методом ГЖХ не превышает 5% относительных.
для идентификации и количественного определения
действующих веществ в дезинсекционных средствах
Определение массовой доли ДВ включает следующие операции: подготовку анализируемой пробы (часто приходится выделять ДВ из некоторых форм применения методом экстракции); идентификацию и количественное определение ДВ методом ГЖХ.
Способы подготовки пробы при анализе различных форм применения. Способы подготовки пробы при анализе ДВ зависят от вида форм применения дезинсекционных средств. В случае безводных жидких форм, в которых растворителями являются органические растворители, или концентрированных водных растворов пробу готовят простым разведением средства. В случае же разбавленных жидких водных растворов, эмульсий, порошков и других твердых форм применения обязательной начальной стадией анализа является выделение ДВ путем экстракции его из анализируемых средств органическими растворителями. При этом степень извлечения ДВ зависит от состава средства - массовой доли ДВ и сопутствующих компонентов. Чем сложнее состав средства и чем ниже концентрация в нем ДВ, тем коэффициент извлечения их ниже.
Способы выделения ДВ из различных форм применения дезинсекционных средств приведены в табл. 4.1.
Таблица 4.1
Способы выделения действующих веществ
из различных форм применения
Форма применения | Действующее вещество, % | Способ подготовки анализируемой пробы |
1 | 2 | 3 |
1. Концентраты эмульсии к.э. | - перметрин - 5 - 25; - циперметрин - 25; - альфа-циперметрин - 10; - фенотрин - 10; - дельтаметрин - 2,5; - фентион - 25; - хлорпирифос - 48; - малатион - 50 - 57; - пропоксур - 20 | Разведение концентрата эмульсии полярным или неполярным растворителем до необходимой концентрации |
2. Водно-спиртовые лосьоны: | Разведение лосьона полярным растворителем, удаление следов воды прокаленным осушителем | |
- педикулицид; | - перметрин - 0,5; | |
- антимольный спиртовой раствор в БАУ; | - перметрин - 0,55; | |
- репеллентный спиртовой раствор в БАУ | - ИР3535 - 12,0 | |
3. Смачивающийся порошок | - альфа-циперметрин - 5,0; - фентион - 40,0; - азаметифос - 50,0 | Экстракция неполярным растворителем при перемешивании на магнитной мешалке в течение 1 - 3 ч; Kизвл = 90 - 95% |
4. Твердые формы: | - перметрин - 13,0 | Измельчение, экстракция неполярным растворителем при перемешивании на магнитной мешалке в течение 1 - 3 ч; Kизвл = 90 - 95% |
- таблетки, шашки; | таблетка, шашка; | |
- дусты; | - перметрин - 0,25 дуст; | |
- мелок, карандаш | - ДЭТА - 17,5 (карандаш); - альфа-циперметрин - 0,3 карандаш | |
5. Аэрозольные баллоны | - пралле - 0,32; - альфа-циперметрин - 0,20; - пропеллент УВП - 50; - цифенотрин - 0,2; - тетраметрин - 0,1; - УВП - 60,0 [40] | Удаление углеводородного пропеллента УВП методом испарения, разведение состава соответствующим растворителем до необходимой концентрации; Kизвл = 85 - 90% |
6. Гели, пасты | - хлорпирифос - 0,6, гель; - ДЭТА - 17,5, крем-гель; - пропоксур - 2,0, приманка | Экстракция неполярным растворителем; удаление следов воды прокаленным осушителем; Kизвл = 80 - 85% |
7. Ткани, импрегнированные составом перметрина | - перметрин - 0,5; - хлорпирифос - 0,6 | Экстракция неполярным растворителем; экстракция неполярным растворителем |
- бумага, пропитанная хлорпирифосом |
Идентификация и количественное определение действующих веществ методом газожидкостной хроматографии. Сущность метода заключается в разделении смеси веществ на хроматографической колонке. Входным сигналом хроматографа является концентрация или масса определяемого компонента на входе в хроматограф.
Выходным сигналом хроматографа являются: амплитуда в максимуме хроматографического пика (высота пика) или площадь хроматографического пика и время удерживания определяемого компонента. По времени удерживания ДВ осуществляется его идентификация; по высоте хроматографического пика или площади рассчитываются концентрация и массовая доля ДВ.
Средства измерения:
- хроматографы лабораторные разных марок (ЛХМ-80, "Цвет", "Кристалл" или другой аналогичный) с пламенно-ионизационным детектором ПИД;
- аналитическая колонка (металлическая или стеклянная) длиной 100 или 200 см и внутренним диаметром 0,3 см;
- наполнитель - хроматон N-AW-HMDS с 5% SE-30 или хромосорб W-HP с 3% OV-210.
Выполнение анализа. Анализ дезинсекционного средства включает следующие операции:
- приготовление градуировочных растворов точной концентрации (0,5 - 2,0 мг/см3) для градуировки хроматографа (построение графика зависимости высоты площади хроматографического пика от концентрации ДВ;
- приготовление раствора анализируемой пробы (разведением или экстракцией средства);
- хроматографирование анализируемого раствора параллельно с градуировочным раствором определенной концентрации в аналогичных условиях.
Условия хроматографирования:
Скорость газа-носителя азота, см3/мин | 30 - 35 |
Скорость водорода, см3/мин | 30 - 35 |
Скорость воздуха, см3/мин | 300 |
Чувствительность шкалы электрометра | 5-10 x 10-10 A |
Концентрация анализируемого и стандартного растворов, мг/см3 | 0,5 - 2,0 |
Температура колонки, °C <*> | 170 - 260 |
Температура детектора, °C | 190 - 270 |
Температура испарителя, °C | 190 - 280 |
--------------------------------
На хроматограммах измеряются высоты или площади хроматографических пиков.
Обработка результатов анализа раздел 4.3.2.
В настоящее время известны около 18 пиретроидов, используемых в качестве ДВ в составе различных дезинсекционных средств. Распределение пиретроидов по назначению: в качестве педикулицидов используют фенотрин и перметрин (0,3 - 0,5%); инсектоакарицидов - циперметрин и альфа-циперметрин (0,20 - 0,25%); инсектицидов с высоким "нокдаун-эффектом" в составе аэрозольных баллонов - тетраметрин (0,1 - 1,0%) и имипротрин (0,1 - 0,2%) в сочетании с другими пиретроидами (перметрин, циперметрин, фенотрин, цифенотрин) или ФОС сумитрин; электрофумигирующих средств - изомеры аллетрина (биоаллетрин, эсбиотрин) и праллетрин (10 - 20 мг/см3); в противомольных средствах - вапортрин, перметрин.
Из пиретроидов отобраны 11 (табл. 4.2.) наиболее востребованных, для 10 из которых зарегистрированы субстанции (технические продукты).
В качестве примера использования метода ГЖХ для количественного определения пиретроидов приведен анализ технического продукта перметрина.
Массовую долю перметрина определяют методом ГЖХ с использованием пламенно-ионизационного детектирования и количественной оценки перметрина методом абсолютной градуировки по стандартному образцу ГСО 7715-99.
Для анализа используют аналитический газовый хроматограф, снабженный ПИД, металлической колонкой длиной 100 см и диаметром 0,3 см, заполненной силанизированным хроматоном N-AW-DMCS 0,20 - 0,25, пропитанным 5% неподвижной фазы SE-30.
С целью определения зависимости высоты (площади) хроматографических пиков от концентрации анализируемых растворов разово готовят и хроматографируют рабочие градуировочные растворы с концентрацией ДВ 0,5; 1,0; 1,5 и 2,0 мг/см3 в четыреххлористом углероде.
После установления линейной зависимости в изучаемой области концентраций анализ перметрина технического проводят следующим образом.
Выполнение анализа. Анализируемый раствор готовят растворением 0,25 г технического продукта в четыреххлористом углероде в мерной колбе вместимостью 50 см3 с последующим разведением аликвоты полученного раствора в 5 раз тем же растворителем.
Приготовленный анализируемый раствор вводят в хроматограф не менее 3 раз параллельно с рабочим градуировочным концентрации 1,0 мг/см3.
Условия хроматографирования:
Температура колонки, °C | 250 |
Температура детектора, °C | 260 |
Температура испарителя, °C | 260 |
Чувствительность шкалы электрометра | 10 x 10-10 A |
Объем вводимой пробы, мкл | 1 |
Время удерживания перметрина | 3 мин 30 с |
Обработка результатов. Массовую долю перметрина X в процентах рассчитывают по формуле:
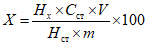 , где
, гдеHx и Hст - высоты хроматографических пиков перметрина в анализируемом и рабочем градуировочном растворах, мм;
Cст - концентрация перметрина в градуировочном растворе, мг/см3;
V - объем анализируемого раствора, см3;
m - масса навески субстанции (перметрина технического), мг.
За результат анализа принимают среднее арифметическое значение из 3 определений, абсолютное расхождение между которыми не превышает 0,5%.
Ниже в табл. 4.2 представлен метод анализа различных форм применения пиретроидов в обобщенном виде. При этом для каждого пиретроида приведены наиболее распространенные формы применения, условия их хроматографирования, метод количественной оценки (абсолютная градуировка с использованием стандартного образца определяемого вещества или метод "внутреннего эталона"), растворитель для разведения или экстракции ДВ из дезинсекционного средства и коэффициент извлечения ДВ.
Таблица 4.2
в различных формах применения
Название ДВ, формы применения | Способ приготовления анализируемой пробы | Условия хроматографирования |
1 | 2 | 3 |
1. ПЕРМЕТРИН 3-Феноксибензил(1RS)-цис,транс-3-(2,2-дихлорвинил)-2,2-диметил-цикло-пропанкарбоксилат | ||
- Субстанция "Перметрин технический" | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; |
не менее 92,0% (соотношение цис-транс-изомеров 25:75) | количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 1,0 мг/см3; температура колонки - 250 °C; температура детектора - 260 °C; температура испарителя - 260 °C; время удерживания перметрина - 3 мин 30 с | |
Формы применения: | ||
- Педикулицидные лосьоны перметрин - 0,5% | Разбавление полярным растворителем до необходимой концентрации, удаление следов воды прокаленным осушителем | |
- Педикулицидные шампуни перметрин - 0,3% | Экстракция гептаном, удаление следов воды прокаленным осушителем | |
- Концентраты эмульсий перметрин - 5 - 25% | Разведение неполярным растворителем до определенной концентрации | |
- Мыло перметрин - 0,5% | Экстракция гептаном | |
- Аэрозольные баллоны перметрин - 0,2% | Удаление пропеллента путем испарения, разведение состава неполярным растворителем до необходимой концентрации | |
- Таблетки, шашки перметрин - 13,0% | Экстракция неполярным растворителем | |
- Растворы, БАУперметрин - 0,55% | Разведение полярным растворителем до необходимой концентрации, удаление следов воды прокаленным осушителем | |
- Кремы перметрин - 1,0% | Экстракция смесью полярного и неполярного растворителей в соотношении 1:4, удаление следов воды прокаленным осушителем | |
2. ЦИПЕРМЕТРИН Альфа-циано-3-феноксибензил 1RS-цис,транс-3-(2,2-дихлорвинил)-2,2-диметилциклопропан-карбоксилат | ||
- Субстанция "Циперметрин технический", не менее 95,0% (соотношение цис-, транс- изомеров 50:50) | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 1,0 мг/см3; температура колонки - 250 °C; температура детектора - 260 °C; температура испарителя - 260 °C; время удерживания циперметрина - 4 мин 30 с |
Формы применения: | ||
- Концентраты эмульсий 25,0% к.э. циперметрина | Разбавление полярным растворителем до необходимой концентрации, удаление следов воды прокаленным осушителем | |
- Смачивающийся порошок циперметрин - 2,9%; малатион - 14,0% | Экстракция неполярным растворителем | |
Твердые формы: дуст (циперметрин - 0,24%), меловой карандаш - циперметрин - 0,24% | Экстракция неполярным растворителем | |
- Аэрозольные баллоны циперметрин - 0,1%; имипротрин - 0,08%; МГК 264 - 0,8%; УВП - 50,0% | Удаление пропеллента путем испарения, разведение состава неполярным растворителем до необходимой концентрации | |
- Гели циперметрин - 0,1% | Экстракция неполярным растворителем | |
3. АЛЬФА-ЦИПЕРМЕТРИН (сумма 2 цисизомеров циперметрина) | ||
- Субстанция "Альфа-циперметрин технический", не менее 95,0% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 1,0 мг/см3; температура колонки - 250 °C; температура детектора - 260 °C; температура испарителя - 260 °C; время удерживания альфа-циперметрина - 4 мин 30 с |
Формы применения: | ||
- Аэрозольные баллоны альфа-циперметрин - 0,15%; перметрин - 0,2% | Удаление пропеллента путем испарения, разведение состава неполярным растворителем | |
- Растворы, БАУ альфа-циперметрин - 0,22% | Разбавление полярным растворителем до необходимой концентрации, удаление следов воды прокаленным осушителем | |
Гели альфа-циперметрин - 0,2% | Экстракция неполярным растворителем | |
4. ДЕЛЬТАМЕТРИН S-альфа-циано-3-феноксибензил (1R, 3R)-3-(2,2-дибромвинил)-2,2-(диметил)-циклопропанкарбоксилат | ||
- Субстанция, содержащая не менее 98,0% основного вещества | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 1,0 мг/см3; температура колонки - 250 °C; температура детектора - 260 °C; температура испарителя - 260 °C; время удерживания дельтаметрина - 6 мин 30 с |
Формы применения: | ||
- Концентраты эмульсий дельтаметрин - 2,5% | Экстракция неполярным растворителем | |
Твердые формы: - Дуст дельтаметрин - 0,025%; фентион - 0,40%; мелокдельтаметрин - 0,1%; зетациперметрин - 0,05% | Экстракция неполярным растворителем | |
- Аэрозольные баллоны дельтаметрин - 0,02% | Удаление пропеллента путем испарения, экстракция из состава неполярным растворителем, удаление следов воды прокаленным осушителем | |
5. ЭСБИОТРИН 1RS-Аллил-2-метил-4-оксо-циклопент-2-енил-(1R, 3R)- 2,2-диметил-3-(2-метил-проп-1-енил)-циклопропанкарбоксилат | ||
- Субстанция "Эсбиотрин", суммарное содержание ДВ в техническом продукте не менее 93,0% | Растворение в полярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по дибутилфталату; растворитель - ацетон; Cст. = Cан. = 1,0 мг/см3; температура колонки - 200 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удержания эсбиотрина - 2 мин 20 с |
Формы применения: | ||
Электрофумигирующие средства: - Электрофумигатор жидкостной эсбиотрин - 3,0% | Разведение полярным растворителем | |
- Электрофумигатор с пластинами от комаров эсбиотрин - 20,0 мг/на пластину | Измельчение, экстракция полярным растворителем | |
- Свеча эсбиотрин - 0,16% | Экстракция полярным растворителем | |
6. ПРАЛЛЕТРИН ЭТОК S-2-метил-4-оксо-3-проп-2-инил-циклопент-2-енил-1R-2,2-диме - диметил-3-(2-метил-проп-1-енил)-циклопропанкарбоксилат | ||
- Субстанция ЭТОК с содержанием действующего вещества в техническом продукте не менее 93,0% | Растворение в полярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по дибутилфталату; растворитель - ацетон; Cст. = Cан. = 1,0 мг/см3; температура колонки - 200 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания праллетрина - 2 мин 20 с |
Формы применения: | ||
- Электрофумигирующее средство в форме пластины содержание праллетрина - 10 мг на пластину | Измельчение пластины, экстракция полярным растворителем | |
7. НЕОПИНАМИН ФОРТЕ (d-ТЕТРАМЕТРИН) 3,4,5,6-Тетрагидро-фталимидометил-1R-цис, транс-хризантемат | ||
- Субстанция содержит минимум 92,0% активных d-транс-изомеров | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по дибутилфталату; растворитель - ацетон; Cст. = Cан. = 1,0 мг/см3; температура колонки - 250 °C; температура детектора - 270 °C; температура испарителя - 260 °C; время удерживания тетраметрина - 2 мин 30 с; программирование температуры от 200 до 250 °C |
Формы применения: | ||
- Аэрозольные баллоны (для летающих и ползающих насекомых) перметрин - 0,2%; циперметрин - 0,2%; тетраметрин - 0,2%; ППБ - 1,0%; УВП - 40,0% | Удаление пропеллента путем испарения, разведение состава неполярным растворителем | |
Твердые формы: - Дуст тетраметрин - 0,05%; перметрин - 0,45%; фенвалерат - 0,15% | Экстракция неполярным растворителем | |
8. ПРАЛЛЕ (50,0% ИМИПРОТРИН) [2,5-Диоко-3-(2-пропинил)-1-имидазолидинил]-метил 1RS-цис, транс-хризантемат | ||
- Субстанция ПРАЛЛЕ (50% имипротрин) | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка методом абсолютной градуировки или методом "внутреннего эталона" по дифенилфталату; растворитель - ацетон; Cст. = Cан. = 1,0 мг/см3; температура колонки - 190 - 260 °C; температура детектора - 260 °C; температура испарителя - 260 °C; время удерживания имипротрина - 2 мин 15 с |
Формы применения: | ||
Аэрозольные баллоны: - Инсектицидное средство имипротрин - 0,1%; d-цифенотрин - 0,3%; УВП - 40,0% | Удаление пропеллента путем испарения, разведение состава полярным растворителем | |
- Акарицидное средство пралле - 0,32%; альфациперметрин - 0,2%; УВП - 50% | Удаление пропеллента путем испарения, разведение состава полярным растворителем | |
9. ГОКИЛАТ-S ЦИФЕНОТРИН RS-Альфа-циано-3-феноксибензил-1R-цис,транс- хризантемат | ||
- Субстанция ГОКИЛАТ-S ЦИФЕНОТРИН - технический продукт с содержанием основного вещества - 96,0% | Растворение в полярном растворителе | Метод ГЖХ с ПИД; количественная оценка методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки, детектора и испарителя - 250 °C; время удерживания цифенотрина - 2 мин 30 с |
Формы применения: | ||
Аэрозольные баллоны - Инсектицидное средство цифенотрин - 0,2%; d-тетраметрин - 0,1%; УВП - 60,0% | Удаление пропеллента путем испарения, разведение неполярным растворителем | |
- Концентрат эмульсии цифенотрин - 10% | Разведение неполярным растворителем до необходимой концентрации | |
10. d-ФЕНОТРИН СУМИТРИН 3-Феноксибензил-1RS-цис, транс-2,2-диметил-3-(2,2-диметилвинил)-циклопропанкарбоксилат | ||
- Технический продукт с содержанием основного вещества 93,7% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по диоктилфталату; растворитель - четыреххлористый углерод; Cст. = Cан. = 1,0 мг/см3; температура колонки - 230 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания d-фенотрина - 4 мин 15 с |
Формы применения: | ||
Аэрозольные баллоны: Инсектицидное средство d-фенотрина - 0,15%; d-тетраметрин - 0,15% | Удаление пропеллента путем испарения, разведение неполярным растворителем | |
Ловушки: - Инсектицидное средство, ловушка от муравьев фенотрин - 0,1%; аттрактант - 98,6% | Экстракция неполярным растворителем | |
- Концентрат эмульсии фенотрин - 10,0% | Разведение неполярным растворителем до нужной концентрации | |
11. ВАПОРТРИН ЭМПЕНТРИН RS-1-3-Этинил-2-метилпент-2-енил-1R-цис, транс- хризантемат | ||
- Субстанция вапортрина технический продукт с содержанием основного вещества не менее 93,0% | Растворение в полярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - ацетон; Cст. = Cан. = 1,0 мг/см3; температура колонки - 170 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания вапортрина - 2 мин 10 с |
Формы применения: | ||
- Инсектицидное средство, в виде бумажных пластин, пропитанных раствором вапортрина; вапортрин - 8,6% | Экстракция полярным растворителем | |
- Инсектицидное средство от мух в виде жидкости с содержанием вапортрина вапортрин - 2,8% | Разведение полярным растворителем |
Основными фосфорорганическими соединениями, используемыми в качестве ДВ в составе различных инсектицидов и акароинсектицидов в настоящее время, являются: хлорпирифос, хлорофос, малатион карбофос, фентион сульфидофос, сумитион (фенитротион), диазинон и азаметифос.
В качестве субстанций зарегистрированы: хлорофос, хлорпирифос и фентион технический сульфидофос.
Анализ хлорофоса осуществляют методом ВЭЖХ, анализ азаметифоса проводится спектрофотометрическим методом при длине волны 295 нм.
Остальные пять фосфорорганических соединений определяют методом ГЖХ/ПИД при температуре колонки 190 - 210 °C, температуре испарителя и детектора 200 - 230 °C.
В качестве внутреннего стандарта используют дибутилфталат или дифенил.
В табл. 4.3 представлены методы количественного определения фосфорорганических соединений в обобщенном виде для различных форм применения.
Подробно условия хроматографирования при использовании метода ГЖХ приведены в п. 4.3.2.
Таблица 4.3
Методы определения фосфорорганических соединений
Действующие вещества, препаративные формы | Способ подготовки анализируемой пробы | Условия хроматографирования |
1 | 2 | 3 |
1. ХЛОРПИРИФОС, O-Диэтил-O-(3,5,6-трихлор-2-пиридинил)-тиофосфат | ||
- Технический продукт субстанция с содержанием основного продукта 97,0% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 180 °C; температура детектора - 190 °C; температура испарителя - 210 °C; время удерживания хлорпирифоса - 3 мин 40 с |
Формы применения: | ||
- Концентрат эмульсии (хлорпирифос - 20%) в форме микрокапсулированной суспензии | Разбавление смесью ацетонитрила и четыреххлористого углерода (2:3), удаление следов воды прокаленным осушителем | |
- 48,0% к.э. хлорпирифоса | Разбавление неполярным растворителем | |
- Гели гель от тараканов хлорпирифос - 0,6% | Экстракция смесью полярного и неполярного растворителей (1:1), удаление следов воды прокаленным осушителем | |
- Инсектицидное средство в виде гелеобразной пасты хлорпирифос - 0,5% | Экстракция полярным растворителем | |
- Приманки, ловушки-приманки от тараканов | Экстракция смесью полярного и неполярного растворителей (1:1) | |
хлорпирифос - 0,6%; | ||
бумага от моли, хлорпирифос - 2,9%; | Экстракция полярным растворителем | |
порошок-приманка хлорпирифос - 2,0%; | Экстракция неполярным растворителем | |
антимоль, хлорпирифос - 0,6 г на ленту | Экстракция неполярным растворителем | |
2. ФЕНТИОН (Сульфидофос) O,O-Диметил-O-(4-метил-тио-м-толил)-тиофосфат | ||
Технический продукт субстанция "Фентион технический" с содержанием основного вещества не менее 90% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 190 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания фентиона - 2 мин 50 с |
Формы применения: | ||
- Концентраты эмульсий 25% к.э. фентиона | Разбавление неполярным растворителем | |
- Смачивающийся порошок 40% с.п. фентиона | Экстракция неполярным растворителем | |
- Гели | Экстракция неполярным растворителем | |
Средство инсектицидное в форме геля фентион - 0,24% альфа-циперметрин - 0,03% | ||
- Твердые формы дуст фентион - 0,4%; дельтаметрин - 0,025% | Экстракция неполярным растворителем | |
3. ХЛОРОФОС O,O-Диметил-(1-окси-2,2,2-трихлорэтил)-фосфонат | ||
- Технический продукт субстанция с содержанием хлорофоса 98,0% | Разбавление элюентом: метанол - 0,3% водный раствор ортофосфорной кислоты 55:45 | Метод ВЭЖХ с УФ-детектированием при длине волны - 220 нм; сорбент - IP, элюент - метанол-ортофосфорная кислота 55:45; количественная оценка - методом абсолютной градуировки |
Инсектицидное средство хлорофос - 80,0% с.п. | Экстракция элюентом | |
4. МАЛАТИОН КАРБОФОС O,O-Диметил-S-[1,2-ди(этоксикарбонил)этилдитио]-фосфат | ||
Формы применения: | ||
- Концентраты эмульсий малатион - 57% к.э.; малатион - 10,0%; циперметрин - 20,0%; - малатион - 50,0% к.э. | Разбавление неполярным растворителем | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 190 °C; температура детектора - 200 °C; температура испарителя - 220 °C; время удерживания малатиона - 2 мин 30 с |
Твердые формы: таблетка малатион - 8,0%; альфа-циперметрин - 20% | Экстракция неполярным растворителем | |
- Инсектицидное средство в виде порошка малатион - 14,0%, циперметрин - 2,9% | Экстракция неполярным растворителем | |
5. ДИАЗИНОН O,O-Диэтил-O-(2-изопропил-(4-метил-пиримидил-6)-тиофосфат | ||
Гели, ловушки: инсектицидное средство в форме таблетки диазинон - 6,0% | Экстракция неполярным растворителем | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 170 °C; температура детектора - 200 °C; температура испарителя - 220 °C; время удерживания диазинона - 2 мин 10 с |
- Инсектицидное средство в форме геля диазинон - 0,2%; хлорпирифос - 0,3% | Экстракция неполярным растворителем | |
- Инсектицидное средство в форме гель-пасты диазинон - 0,3%; альфа-циперметрин - 0,05% | Экстракция неполярным растворителем | |
- Инсектицидное средство в форме геля диазинон - 0,225%; циперметрин - 0,075% | Экстракция полярным растворителем | |
6. ФЕНИТРОТИОН СУМИТИОН O,O-Диметил-O-(4-нитро-м-толил)-тиофосфат | ||
Формы применения: | ||
Аэрозольные баллоны: - Аэрозольный баллон для нелетающих насекомых фенитротион - 1,0%; тетраметрин - 0,1%; УВП - 55% | Удаление пропеллента путем испарения, разбавление неполярным растворителем | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 200 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания фенитротиона - 2 мин 3 с |
- Аэрозольный баллон для летающих насекомых тетраметрин - 0,075%; перметрин - 0,04%; фенитротион - 0,3%; синергист - пиперонилбутоксид ППБ - 0,3%; УВП-бутан - 67,5% | Удаление пропеллента путем испарения, разбавление неполярным растворителем | |
7. АЗАМЕТИФОС O,O-диметил-S-(2-оксо-6-хлор-пиридооксазолил-3-метил)-тиофосфат | ||
- Инсектицидное средство "Альфакрон 50% с.п." | Экстракция неполярным растворителем | |
- Инсектицидное средство в форме порошка альфакрон - 0,4% или азаметифос - 0,2% | Экстракция неполярным растворителем | |
Приманка для мух в виде пластины азаметифос - 1,0 мг на пластину | Экстракция неполярным растворителем | Спектрофотометрический метод (длина волны - 295 нм) |
За последние 5 лет нашли применение средства с двумя ДВ из класса карбаматов - метомилом и пропоксуром.
В нижеследующей табл. 4.4 приведены методики количественного определения указанных ДВ в средствах на основе карбаматов.
Таблица 4.4
Методики определения карбаматов
Действующие вещества, формы применения | Способ подготовки анализируемой пробы | Условия хроматографирования |
1 | 2 | 3 |
1. МЕТОМИЛ 1-Метилтиоацетальдегида O-(N-метил-карбамоил)оксим | ||
- Технический продукт Субстанция "Метомил" с содержанием основного вещества не менее 98,0% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 100 °C; температура детектора - 200 °C; температура испарителя - 200 °C; время удерживания метомила - 2 мин 20 с |
- Приманка от мух Инсектицидное средство в виде гранулированной приманки для мух метомил - 1,0% | Экстракция неполярным растворителем | |
2. ПРОПОКСУР O-(2-Изопропокси-карбонилпропен-1-ил-2)-O-метил-N-этиламидотиофосфат | ||
- Концентрат эмульсии (пропоксур, 20% к.э.) | Разведение полярным растворителем | Метод ГЖХ с ПИД; количественная оценка методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 180 °C; температура детектора - 230 °C; температура испарителя - 230 °C; время удерживания пропоксура - 2 мин 20 с |
- Приманка для уничтожения тараканов (пропоксур - 2,0%) | Экстракция полярным растворителем |
Подробно условия хроматографирования приведены в п. 4.3.2.
размножения тараканов - S-гидропрена
Метод ГЖХ для определения регулятора размножения тараканов - S-гидропрена представлен в табл. 4.5.
Таблица 4.5
Метод ГЖХ для определения регулятора размножения
тараканов S-гидропрена
Действующие вещества, формы применения | Способ подготовки анализируемой пробы | Условия хроматографирования |
S-Гидропрен | ||
- Средство инсектицидное регулятор размножения тараканов "S-Гидропрен" с содержанием основного вещества не менее 99,1% | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - гексан; Cст. = Cан. = 2,0 мг/см3; температура колонки - 200 °C; температура детектора - 250 °C; температура испарителя - 250 °C; время удерживания S-гидропрена - 3 мин 5 с |
Подробно условия хроматографирования приведены в п. 4.3.2.
действующих веществ в репеллентных средствах
Основными репеллентами, широко применяемыми в сфере медицинской дезинсекции, являются диметилфталат ДМФ, N,N-диэтил-мета-толуамид ДЭТА, N-гексилоксиметилкапролактам акреп, этил-3-N-бутилацетамино-пропионат (репеллент ИР3535), 1-(1-метилпропокси-карбонил)-2-(2-гидроксиэтилен)-пиперидин (КБР).
Основные формы применения - аэрозольные баллоны, растворы в беспропеллентной упаковке БАУ, лосьоны, эмульсии, кремы, карандаши, салфетки.
Наиболее востребованным репеллентом является ДЭТА. В составе аэрозольных баллонов, предназначенных для уничтожения кровососущих насекомых, его содержание - 10,0 - 20,0%, в составе аэрозольных баллонов, предназначенных для защиты от клещей - 30 - 32%. Часто ДЭТА комбинируют с другими репеллентами, например в составе лосьонов с ДМФ, в сочетании с репеллентом ИР3535 в спреях или в сочетании с альфа-циперметрином в акароинсектицидных средствах в форме аэрозольных баллонов.
Акреп - репеллент отечественного производства - используется для изготовления эмульсий, как правило, в сочетании с ДМФ.
В качестве субстанций в настоящее время зарегистрированы репеллент ИР3535 производства фирмы "Мерк" Германия и ДЭТА производства фирмы "МакЛафлин Гормли Кинг" США.
Анализ всех репеллентов осуществляют методом ГЖХ на хроматографе с ПИД и количественной оценкой ДВ методом абсолютной градуировки или методом "внутреннего эталона" по дипропилфталату или по дифенилу.
Основная задача при анализе различных репеллентных средств заключается в количественном выделении ДВ из препаративной формы путем подбора оптимальных условий выделения: растворителя для экстракции ДВ, времени экстракции, температурного режима. Наименьший коэффициент извлечения (85 - 90%) достигается при анализе кремов, что обусловлено их сложным составом.
В табл. 4.6 представлен анализ репеллентов в обобщенном виде для различных препаративных форм. Для каждого репеллента приведены наиболее распространенные формы применения, ДВ в препаративной форме, условия хроматографирования, растворитель для экстракции и коэффициент извлечения ДВ.
Таблица 4.6
Газохроматографический метод анализа репеллентов
в различных формах применения
Название ДВ, формы применения | Способ подготовки анализируемой пробы | Условия хроматографирования |
1 | 2 | 3 |
1. ДМФ Диметилфталат | ||
Формы применения: | ||
- Репеллентное средство лосьон (ДМФ - 30%) | Разведение этиловым спиртом | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки [18] или методом "внутреннего эталона" по дециловому спирту; растворитель - этиловый спирт; Cст. = Cан. = 2,0 мг/см3; температура колонки - 160 °C; температура детектора - 170 °C; температура испарителя - 160 °C; время удерживания ДМФ - 2 мин 30 с |
- Репеллентное средство в виде водно-спиртового раствора в БАУ ДЭТА - 14,0%; ДМФ - 9,0% | Разведение этиловым спиртом | |
- Репеллентное средство в форме эмульсии ДМФ - 20,0%; акреп - 20,0% | Разведение этиловым спиртом | Количественная оценка методом "внутреннего эталона" по дипропилфталату; время удерживания ДМФ - 3 мин 30 с; время удержания акрепа - 5 мин 55 с |
2. ДЭТА N,N-диэтил-мета- толуамид | ||
Формы применения: | ||
Репеллентное средство в аэрозольной упаковке ДЭТА - 25,0% | Удаление пропеллента путем испарения, разбавление состава растворителем | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по дибутилфталату; растворитель - этиловый спирт; Cст. = Cан. = 2,0 мг/см3; температура колонки - 170 °C; температура детектора - 180 °C; температура испарителя - 190 °C; время удерживания ДЭТА - 3 мин 30 с |
- Инсектицидно-репеллентное средство в аэрозольной упаковке ДЭТА - 15,0%; альфа-циперметрин - 0,18% | Удаление пропеллента путем испарения, разбавление состава растворителем | |
- Инсектоакарицидное средство (ДЭТА - 30,0%) в форме аэрозольного баллона | ||
- Репеллентное средство в форме водно-спиртового раствора БАУ ДЭТА - 10,0% | Разведение полярным растворителем | |
- Репеллентное средство в виде карандаша ДЭТА - 17,5% | Экстракция полярным растворителем | |
- Средство репеллентное в виде карандаша ДЭТА - 15,0% | Экстракция этиловым спиртом | |
- Репеллентное средство в виде крем-геля ДЭТА - 17,5% | Экстракция смесью ацетон:четыреххлористый углерод (1:1) | |
3. ИР3535 Этил-3-(N-бутил-ацетамидо)-пропионат | ||
Субстанция "ИР 3535" - технический продукт, содержащий не менее 98,0% основного вещества | Растворение в неполярном растворителе | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - четыреххлористый углерод; Cст. = Cан. = 2,0 мг/см3; температура колонки - 150 °C; температура детектора - 210 °C; температура испарителя - 210 °C; время удерживания ИР3535 - 2 мин 30 с |
Формы применения: | ||
- Репеллентное средство антикомариный спрей для детей, в форме БАУ ИР3535 - 12,0% | Разбавление этиловым спиртом | |
- Репеллентное средство антикомариный спрей для взрослых ИР3535 - 4,0%; ДЭТА - 5,0%; гександиол - 5,0% | Разбавление этиловым спиртом | Температура колонки - 130 - 160 °C; температура детектора и испарителя - 230 °C; время удерживания: гександиола - 2 мин 20 с; ДЭТА - 6 мин 23 с; ИР3535 - 6 мин 45 с |
4. КБР 1-Пиперидинкарбоновая кислота, 2-(2-гидроксиэтил)-1-метилпропиловый эфир | ||
Формы применения: | ||
Лосьоны: - Репеллентное средство лосьон в форме водно-спиртового раствора БАУ КБР - 20,0% | Разведение этиловым спиртом | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки; растворитель - этиловый спирт; Cст. = Cан. = 2,0 мг/см3; температура колонки - 160 °C; температура детектора - 200 °C; температура испарителя - 200 °C; время удерживания КБР - 3 мин 50 с |
- Репеллентное средство молочко для защиты от летающих насекомых в форме эмульсии КБР - 10,0% | Разведение этиловым спиртом | |
Крем: - Репеллентное средство крем для защиты от летающих насекомых КБР - 10,0% | Разведение этиловым спиртом | |
5. АКРЕП N-(Гексилоксиметил)-капролактам | ||
Формы применения: | ||
Репеллентное средство в форме эмульсии акреп - 22,0%; ДМФ - 18,0% | Экстракция неполярным растворителем | Метод ГЖХ с ПИД; количественная оценка - методом абсолютной градуировки или методом "внутреннего эталона" по дипропилфталату или по дифенилу; растворитель - этиловый спирт; Cст. = Cан. = 2,0 мг/см3; температура колонки - 160 - 190 °C; температура детектора - 230 °C; температура испарителя - 230 °C; время удерживания акрепа - 5 мин 55 с Метод "внутреннего эталона" по дифенилфталату Kизвл = 0,90 - 0,95 |
Подробно условия хроматографирования приведены в п. 4.3.2.
В средствах для медицинской дератизации применяют действующие вещества ДВ различной химической природы и различного механизма действия. В настоящее время в России зарегистрированы родентицидные ДВ, представляющие собой яды острого типа действия и яды пролонгированного действия - антикоагулянты индандионового и кумаринового ряда.
Острые яды, такие как фосфид цинка, глифтор и нафтилтиомочевина, имеют ограниченное применение. Наибольшее применение получили ДВ пролонгированного действия - антикоагулянты индандионового (дифенацин, этилфенацин, изопропилфенацин, хлорфасинон) и кумаринового ряда (зоокумарин и его натриевая соль, бромадиолон, бродифакум, дифетиалон, дифенакум, куматетралил).
Дератизационные средства выпускают в различных препаративных формах: в виде ДВ (индивидуальных и смесевых) с содержанием основного вещества от 30 до 99,8%, жидких и диспергированных твердых порошки концентратов с содержанием ДВ от 0,25 до 2,5%, а также в виде отравленных приманок на пищевой основе с содержанием ДВ от 0,0025 до 3%.
Специфическими требованиями к выпускаемым препаративным формам дератизационных средств являются обязательное добавление интенсивных красителей и горечи (бензоат натрия, битрекс), которые обеспечивают отличие отравленных приманок от пищевых продуктов и предохраняют от случайного отравления на этапах производства и применения дератизационных средств. Исключение составляют фосфид цинка и 1-нафтилтиомочевина, собственная окраска которых не требует дополнительного предупредительного окрашивания.
Основными показателями, которые позволяют идентифицировать средство, являются внешний вид, цвет и содержание ДВ. Внешний вид и цвет определяют визуально. Выбор метода определения ДВ зависит от его физико-химических свойств и уровня содержания, а также состава инертных компонентов в рецептуре средства.
Разнообразие физико-химических свойств ДВ, применяемых в дератизационных средствах, обусловливает возможность применения различных методов их определения: химических - титрование и физико-химических инструментальных методов - спектрофотометрия, газовая хроматография, высокоэффективная жидкостная хроматография.
в дератизационных субстанциях и средствах
Фосфид цинка Zn3P2 представляет собой порошок темно-серого цвета. Действующим веществом является фосфин (фосфористый водород) - PH3, выделяющийся при гидролизном расщеплении фосфида цинка. В качестве примесей технический фосфид цинка может содержать металлический цинк, оксиды цинка и оксиды железа.
Анализ ДВ в техническом продукте фосфида цинка проводят объемным волюмометрическим методом [27] или титриметрическим методом. Указанные методы применимы для анализа фосфида цинка и в препаративных формах дератизационных средств.
Поскольку ратицидная активность фосфида цинка определяется именно токсичностью фосфина, образующегося при кислотном гидролизе фосфида цинка, его определение является предпочтительным для оценки качества дератизационного средства.
Объемный волюмометрический метод определения фосфида цинка основан на кислотном гидролизе фосфида цинка и измерении объема выделившегося фосфина с применением водного раствора сернокислой меди.
Содержание ДВ может быть охарактеризовано по фосфидному фосфору или по фосфидному цинку, которые вычисляют по результатам измерения объема фосфина.
Метод избирателен, примеси в техническом продукте и другие компоненты рецептурного состава в дератизационных средствах не мешают определению.
Приборы, реактивы и растворы. Специальный прибор для измерения объема выделившегося фосфина, в котором последовательно соединены капельная воронка вместимостью 50 мл, круглодонная колба вместимостью 25 - 30 мл, измерительная ячейка вместимостью 100 мл, снабженная водяной рубашкой и уравнительным сосудом с затворной жидкостью - раствором хлористого натрия; поглотительный сосуд с раствором сернокислой меди; весы аналитические; 20%-й раствор соляной кислоты; 5%-й раствор сернокислой меди - поглотительная жидкость; насыщенный раствор хлористого натрия - затворная жидкость; азот газообразный; вода дистиллированная.
Выполнение анализа. Масса средства для анализа берется из расчета 20 - 80 мг фосфидного фосфора или 80 - 400 мг фосфидного цинка в пробе. Анализируемую пробу средства, взвешенную в граммах с точностью до четвертого десятичного знака (около 0,15 г фосфида цинка или около 4 г зерновой приманки), помещают в круглодонную колбу, присоединяют к коммуникациям прибора и, заполнив измерительную бюретку затворной жидкостью, продувают слабым потоком азота в течение двух минут, после чего в капельницу при закрытом кране наливают около 20 мл раствора соляной кислоты. Затем по каплям подают раствор в круглодонную колбу, содержащую анализируемое средство, для разложения фосфида цинка. Постепенно нагревают содержимое колбы до кипения. По окончании реакции газ, оставшийся в колбе, вытесняют в измерительную бюретку насыщенным раствором хлористого натрия. После охлаждения газа в бюретке замеряют его объем. Затем с помощью прибора проводят поглощение фосфина раствором сернокислой меди. Объем выделившегося фосфина определяют по разности объема газа до и после его поглощения.
Обработка результатов. Измеренный объем фосфина приводят к нормальным условиям (V0, мл) по формуле:
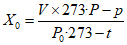 , где
, гдеV - объем фосфина, измеренный в опыте, мл;
P - атмосферное давление, Па (мм рт. ст.);
p - упругость водяных паров над насыщенным раствором хлористого натрия, Па (мм рт. ст.);
P0 - нормальное атмосферное давление, равное 10325 Па (760 мм рт. ст.);
t - температура газа, °C.
Массовую долю фосфина в средстве (X) в процентах вычисляют по формуле:
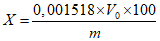 , где
, где0,001518 - масса фосфина, содержащаяся в 1 мл газообразного фосфина, г;
m - масса средства, взятая для анализа, г.
Для того, чтобы вычислить массовую долю фосфидного фосфора в техническом продукте, измеренный объем фосфина пересчитывают на фосфидный фосфор (Xфф) в процентах по формуле:
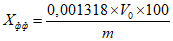 , где
, где0,001518 - масса фосфина, содержащаяся в 1 мл газообразного фосфина, г;
m - масса средства, взятая для анализа, г.
Массовую долю фосфидного цинка (Xфц) в процентах вычисляют из соотношения:
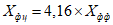 , где
, где4,16 - масса фосфида цинка, соответствующая 1 г фосфидного фосфора, определяемая из отношения молекулярной массы фосфида цинка к массе двух атомов фосфора 258,06 г / 2 x 31 г.
Титриметрический метод определения фосфидного цинка в техническом продукте субстанции [27] основан на раздельном титровании общего и свободного металлического цинка. Содержание фосфидного цинка определяют по разности полученных результатов.
Приборы, реактивы и растворы. Весы аналитические; бюретка и стеклянная мерная посуда; точно 0,1 н раствор тиосульфата натрия; 10%-й раствор йодистого калия; 1 М раствор гидроокиси натрия; аммиачно-хлоридный буфер с pH 10,0 - 10,5; 0,1 М раствор уксусной кислоты; 2 М раствор серной кислоты; 0,1 М раствор сернокислой меди; точно 0,1 н раствор трилона Б; 1%-й раствор крахмала индикатор; эриохром черный Т индикатор.
Выполнение анализа.
Определение металлического цинка.
В мерную колбу вместимостью 25 см3 вносят навеску фосфида цинка массой 0,1 г, взвешенную с точностью до 0,0002 г, или эквивалентное количество отравленной приманки. Приливают 5 см3 0,1 М раствора сульфата меди. Смесь оставляют на 30 мин, периодически встряхивая. Затем раствор отфильтровывают через бумажный фильтр, осадок промывают на фильтре дважды по 5 см3 дистиллированной воды, приливают к фильтрату 1 см3 0,1 М уксусной кислоты, 5 см3 10%-го раствора йодистого калия. Выделившийся йод титруют 0,1 н раствором тиосульфата натрия до слабо-желтой окраски. Аналогично титруют 5 см3 0,1 М раствора сульфата меди после прибавления 1 см3 0,1 М уксусной кислоты и 5 см3 10%-го раствора йодистого калия - холостую пробу.
Обработка результатов. Массовую долю металлического цинка в средстве Zn, % вычисляют по формуле:
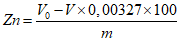 , где
, где0,00327 - масса цинка, соответствующая 1 мл точно 0,1 н раствора тиосульфата натрия, г;
V0 - объем 0,1 н раствора тиосульфата натрия, израсходованный на титрование холостой пробы, см3;
V - объем 0,1 н раствора тиосульфата натрия, израсходованный на титрование холостой пробы, см3;
m - масса средства, взятая на анализ, г.
Определение массовой доли основного вещества. К навеске технического образца фосфида цинка массой 0,1 г, взвешенной с точностью до 0,0002 г, или к эквивалентному количеству отравленной приманки прибавляют 5 - 10 см3 5 М серной кислоты, 25 см3 дистиллированной воды и кипятят в течение 15 мин. Смесь охлаждают и нейтрализуют 1 М раствором гидроксида натрия до pH 3 - 5, приливают 5 см3 аммиачно-хлоридного буфера с pH 10 - 10,5 и титруют раствором трилона Б с применением индикатора эриохрома черного Т до перехода окраски от красно-фиолетовой до синей.
Обработка результатов. Содержание фосфидного цинка в средстве (Zn3P2) в процентах определяют по формуле:
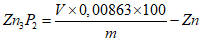 , где
, где0,00863 - масса Zn3P2, соответствующая 1 см3 точно 0,1 н раствора трилона Б, г;
Zn - массовая доля металлического цинка в пробе, определенная при титровании тиосульфатом натрия, %;
V - объем 0,1 н раствора трилона Б, израсходованный на титрование, см3;
m - масса средства, взятая на анализ, г;
(V x 0,00863 x 100/m) - суммарное содержание фосфидного и металлического цинка, найденное при титровании тиосульфатом натрия, %.
в дератизационных субстанциях и средствах
1-1-нафтил-2-тиомочевины
Определение 1-1-нафтил-2-тиомочевины НТМ спектрофотометрическим методом по собственному поглощению в ультрафиолетовой области спектра основано на измерении оптической плотности растворов НТМ при длине волны в диапазоне 280 - 300 нм.
Определению могут мешать вещества, поглощающие в указанном волновом диапазоне. Метод может использоваться при серийном анализе в условиях производства.
Приборы и реактивы. Для выполнения анализа применяется спектрофотометр СФ-26, СФ-46 или другой марки; стандартная стеклянная мерная посуда; весы аналитические; этиловый спирт 96%-й.
Выполнение анализа. Подготовка пробы к спектрофотометрическому измерению включает растворение технического продукта в этиловом спирте для последующего фотометрирования. Из препаративной формы средства в виде приманки проводят извлечение ДВ 96%-м этиловым спиртом при повышенной температуре (60 °C) в течение 15 мин. Оптическую плотность приготовленного раствора измеряют при длине волны 285 нм в кювете с толщиной поглощающего слоя 1 см. В качестве раствора сравнения используют экстракт, полученный в таких же условиях из "холостой" пробы (не содержащей НТМ).
Обработка результатов. Массовую долю ДВ в средстве X в процентах вычисляют по формуле:
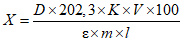 , где
, гдеD - оптическая плотность раствора;
202,3 - молекулярная масса НТМ;
K - кратность разведения экстракта;
V - объем экстракта, л;
m - масса анализируемой пробы, г;
l - толщина поглощающего слоя, см.
Применение справочного значения молярного коэффициента поглощения НТМ при длине волны 285 нм позволяет исключить построение калибровочного графика.
Для определения НТМ в дератизационных средствах применяют также спектрофотометрический метод, основанный на измерении оптической плотности раствора азосоединения, образующегося при взаимодействии НТМ и сульфанилата диазония, при длине волны 490 нм.
Метод избирателен.
Приборы, реактивы и растворы. Для выполнения анализа применяется спектрофотометр СФ-26, СФ-46 или электрофотоколориметр КФК-2; стандартная стеклянная мерная посуда, весы аналитические; этиловый спирт 96%-й, аналитический стандарт НТМ; раствор сульфанилата диазония с массовой концентрацией 5 мг/мл; 0,1 М раствор соляной кислоты; 0,1 М раствор натрия гидроокиси.
Выполнение анализа. При анализе дератизационного средства в виде приманки ДВ извлекают 96%-м этиловым спиртом при повышенной температуре (60 °C) и вносят в аликвоту экстракта реактивы, необходимые для получения азосоединения: раствор НТМ с концентрацией 0,1 мг/мл; раствор сульфанилата диазония с концентрацией 5 мг/мл и 0,1 М раствор соляной кислоты. Смесь выдерживают в течение 5 мин, затем добавляют 0,1 М раствор гидроокиси натрия и фотометрируют при длине волны 490 нм относительно "холостого" раствора калибровочного графика. Для построения калибровочного графика готовят калибровочные растворы азосоединения, эквивалентные содержанию в пробе 20, 40 и 60 мкг НТМ.
Обработка результатов. Массовую долю действующего вещества X в процентах вычисляют по формуле, используя калибровочный график:
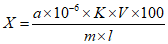 , где
, гдеa - содержание НТМ в аликвоте, найденное по калибровочному графику, мкг;
10-6 - коэффициент пересчета из мкг в г;
K - кратность разведения пробы;
m - масса средства, взятая на анализ, г;
l - толщина поглощающего слоя, см.
1-1-нафтил-2-тиомочевины
Анализ НТМ в зерновой приманке проводят методом высокоэффективной жидкостной хроматографии с применением спектрофотометрического детектирования, градиентного режима хроматографирования и использованием абсолютной градуировки.
Метод избирателен, компоненты рецептурного состава средства не мешают определению.
Приборы, реактивы и растворы. Аналитический жидкостный хроматограф, снабженный спектрофотометрическим детектором, градиентной системой, инжектором с дозирующей петлей 5 - 10 мкл, программой обработки хроматографических данных на базе персонального компьютера; весы аналитические; магнитная мешалка; ротационный испаритель; стандартная стеклянная мерная посуда, НТМ - аналитический стандарт; рабочая градуировочная смесь НТМ в ацетонитриле с массовой концентрацией 0,1 мг/мл; ацетонитрил градации для жидкостной хроматографии; хлористый метилен ч.д.а.; вода бидистиллированная.
Выполнение анализа. Подготовка пробы к анализу предусматривает извлечение ДВ из средства хлористым метиленом в течение 4 ч с помощью магнитной мешалки. Из отфильтрованного экстракта под разрежением отгоняют растворитель, сухой остаток растворяют в ацетонитриле и хроматографируют. Предварительно хроматографируют рабочую градуировочную смесь. Из полученных хроматограмм определяют удерживаемый объем и площади хроматографических пиков НТМ в градуировочной смеси и в анализируемом растворе.
Хроматографирование проводят на колонке для обращенно-фазной хроматографии типа ACCUBOND ODS 5 мкм. В качестве элюентов используют ацетонитрил и воду. Градиент по ацетонитрилу от 40 до 100%. Длина волны измерений 290 нм.
Обработка результатов. Массовую долю НТМ в средстве X в процентах вычисляют по формуле:
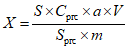 , где
, гдеS и Sргс - площади хроматографических пиков определяемого вещества в анализируемой пробе и рабочей градуировочной смеси;
Cргс - массовая концентрация аналитического стандарта определяемого вещества в рабочей градуировочной смеси;
a - массовая доля основного вещества в аналитическом стандарте, %;
V - объем анализируемого раствора, мл;
m - масса средства, взятая на анализ, мг.
субстанциях и средствах
Особенности химико-аналитических исследований дератизационных ДВ субстанций для производства препаративных форм определяются их составом: содержанием основного вещества, уровнем примесей и содержанием технологических добавок. Для анализа применяют титрование раствором гидроокиси натрия, спектрофотометрические методы с прямым фотометрированием определяемого вещества и в виде окрашенных комплексов, высокоэффективную жидкостную хроматографию со спектрофотометрическим детектированием в ультрафиолетовой области спектра.
этилфенацина и изопропилфенацина
Определение содержания ДВ в субстанциях дифенацина, этилфенацина и изопропилфенацина и концентратах титриметрическим методом [41] основано на титровании индандиона раствором гидроокиси натрия в среде органического растворителя (диоксан; этанол, ацетон) с добавлением индикатора (бромтимоловый синий, феноловый красный) или с применением потенциометрических измерений.
Метод не избирателен, определению могут мешать вещества, титруемые гидроокисью натрия, в связи с этим метод не применим для определения действующего вещества в таких технических продуктах, как трифенацин, тетрафенацин.
Метод может быть рекомендован для рутинных анализов в условиях производства.
Приборы, реактивы и растворы. Весы аналитические; бюретка вместимостью 25 мл; стандартная стеклянная мерная посуда; диоксан; 0,1 М раствор гидроокиси натрия; спиртовой раствор бромтимолового синего индикатор; иономер универсальный ЭВ-74 со стеклянным и каломельным электродами.
Выполнение анализа. Анализируемую пробу растворяют в диоксане и титруют с помощью раствора гидроокиси натрия в присутствии индикатора бромтимолового синего с визуальным или потенциометрическим определением конца титрования.
Обработка результатов. Массовую долю определяемого вещества X в процентах вычисляют по известной формуле:
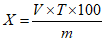 , где
, гдеV - объем точно 0,1 М раствора гидроокиси натрия, израсходованный на титрование пробы, мл;
T - масса определяемого вещества, соответствующая 1 мл точно 0,1 М раствора гидроокиси натрия, г/мл;
m - масса анализируемой пробы, г.
дифенацина, этилфенацина и изопропилфенацина
Определение дифенацина, этилфенацина, изопропилфенацина, хлорфасинона в дератизационных субстанциях (технический продукт, жидкие и диспергированные твердые концентраты) и в препаративных формах проводят спектрофотометрическим методом с прямым определением вещества или в виде окрашенных комплексов.
Спектрофотометрический метод прямого определения изопропилфенацина основан на измерении оптической плотности индан-1,3-диона, в растворе органического растворителя при длине волны 325 и 340 нм относительно растворителя с применением градуировочного графика или молярных коэффициентов поглощения.
Метод избирателен в отсутствии компонентов рецептурного состава средства, поглощающих в указанной области длин волн. Не избирателен в присутствии нескольких веществ индандионового ряда, которые определяются суммарно.
Метод может быть применен для серийного анализа в условиях производства.
Приборы, реактивы и растворы. Для проведения анализа используют спектрофотометр типа СФ-46; стеклянную мерную посуду; этиловый спирт.
Выполнение анализа. Подготовка пробы к анализу заключается в выделении ДВ в раствор. Способ выделения определяемого вещества зависит от рецептурного состава средства. Технический продукт растворяют; из концентратов и приманок ДВ извлекают жидкостной экстракцией с применением этилового спирта и фотометрируют на спектрофотометре при двух длинах волн.
Обработка результатов. Содержание индан-1,3-диона определяют, используя калибровочный график в координатах: оптическая плотность - содержание вещества, мг.
При использовании значений молярных коэффициентов поглощения определяемого индан-1,3-диона содержание ДВ X в процентах вычисляют по формуле:
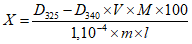
(D325 - D340) - разница оптической плотности анализируемого раствора при разных длинах волн;
M - молекулярная масса определяемого индан-1,3-диона, г/моль;
V - объем раствора или экстракта, л;
1,10-4 - разность молярных коэффициентов поглощения индан-1,3-диона при 325 и 340 нм, л/моль·см.
m - масса анализируемой пробы, г;
l - толщина поглощающего слоя, см.
Спектрофотометрический метод определения дифенацина, этилфенацина и изопропилфенацина в виде окрашенных комплексов основан на образовании водорастворимых окрашенных комплексов ионов железа III с индандионами и измерении их оптической плотности при длине волны 490 нм с применением калибровочного графика или молярных коэффициентов поглощения.
Метод избирателен в отношении традиционно применяемых красителей и других экстрагируемых компонентов рецептурного состава средства. Не избирателен в присутствии нескольких веществ индандионового ряда, которые определяются суммарно.
Метод может быть применен для серийного анализа в условиях производства.
Приборы, реактивы и растворы. Для проведения анализа используют электрофотоколориметр типа КФК или спектрофотометр типа СФ-46; стеклянную мерную посуду; диоксан; 6%-й раствор железа треххлористого.
Выполнение анализа. Подготовка пробы к анализу заключается в выделении ДВ в раствор. Способ выделения определяемого вещества зависит от рецептурного состава средства. Технический продукт растворяют; из концентратов и приманок действующие вещества извлекают жидкостной экстракцией с применением диоксана.
Навеску технического продукта из расчета получения молярной концентрации раствора 2 - 4·10-4 М растворяют в смешивающемся с водой растворителе (диоксан, ацетон) с добавлением небольшого количества воды (до 5%) и 6%-го водного раствора FeCl3·6H2O. Оптимальная концентрация FeIII 2·10-3 М. Раствор фотометрируют при 490 нм. Содержание определяемого индан-1,3-диона вычисляют, используя калибровочный график или молярный коэффициент поглощения комплекса определяемого индан-1,3-диона с железом.
Обработка результатов. Содержание индан-1,3-диона определяют, используя калибровочный график в координатах: оптическая плотность - содержание вещества, мг.
При использовании значений молярных коэффициентов поглощения комплекса определяемого индан-1,3-диона с железом содержание действующего вещества X в процентах вычисляют по формуле:
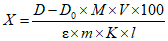
(D - D0) - разница оптической плотности анализируемого и контрольных растворов;
M - молекулярная масса определяемого индандиона, г/моль;
V - объем раствора или экстракта, л;
m - масса пробы, взятой на анализ, г;
K - коэффициент, учитывающий разбавление фотометрируемого раствора водой и солью железа;
l - толщина поглощающего слоя, см.
Экспериментально установлены значения молярных коэффициентов поглощения комплекса железа III для некоторых веществ: дифенацин 1300 +/- 60; хлорфасинон 1260 +/- 80; изопропилфенацин 1100 +/- 80; этилфенацин 960 +/- 80.
антикоагулянтов индандионового ряда
Определение антикоагулянтов индан-1,3-дионового ряда (дифенацин, изопропилфенацин, хлорфасинон, этилфенацин) в субстанциях (технические продукты, жидкие и порошковые концентраты) и различных препаративных формах дератизационных средств проводят методом прямой или обращенно-фазной высокоэффективной хроматографии с применением спектрофотометрического детектирования. Для идентификации и хроматографических измерений антикоагулянтов индан-1,3-дионового ряда применяют спектрофотометрические детекторы на диодной матрице для измерений в области 190 - 400 нм и детекторы для измерений при одной длине волны. Надежность идентификации ДВ, характеризующихся близкими спектрами поглощения в ультрафиолетовой области, достигается использованием двух критериев: 1) спектр поглощения в области 190 - 400 нм; 2) время удерживания в заданных условиях хроматографических измерений.
При анализе технических продуктов (трифенацин, тетрафенацин), представляющих собой малоочищенную технологическую смесь, в которой содержится несколько индандионов с разным содержанием, применение диодно-матричного детектора позволяет записать спектры поглощения каждого пика хроматограммы в диапазоне 190 - 400 нм и идентифицировать индандионы по спектрам поглощения в рабочей градуировочной смеси и испытуемой пробе.
Специфические условия подготовки пробы для анализа обуславливаются содержанием действующего вещества и свойствами ингредиентов рецептуры средства (технологические и функциональные добавки, в т.ч. натуральные пищевые аттрактанты).
Технические продукты и жидкие концентраты растворяют в растворителе, совместимом с хроматографической подвижной фазой. Из порошковых концентратов и пищевых приманок требуется выделение действующего вещества в органическую фазу с помощью экстракции. При этом из-за низкого уровня содержания ДВ в пищевых приманках на фоне сложного рецептурного состава и пищевой основы повышаются требования к селективному выделению действующего вещества и условиям эффективного хроматографического разделения компонентов пробы.
Хроматографический метод определения индандионов с применением прямофазной высокоэффективной хроматографии основан на применении УФ-детектирования, хроматографировании пробы на аминной колонке в изократическом режиме с использованием абсолютной градуировки. В качестве элюента (подвижной фазы) применяют смесь ацетонитрил:вода в соотношении 80:20 по объему.
Избирательность метода. Метод избирателен в присутствии нескольких ДВ индан-1,3-дионового ряда. Определению не мешают технологические примеси в техническом продукте.
Приборы. Аналитический жидкостный хроматограф "Стайер" или другого типа, снабженный спектрофотометрическим детектором на диодной матрице или детектором для измерений при одной длине волны в ультрафиолетовой области спектра, термостатируемой колонкой, изократической системой микронасосов, инжектором типа "Реодайн" (или другого типа) с дозирующей петлей объемом от 5 до 20 мкл, программой управления оборудованием и обработки хроматографических данных на базе персонального компьютера; хроматографическая колонка, как правило, заводского заполнения сорбентом, с соответствующей предколонкой; микрошприц вместимостью 100 - 250 мкл для ручного ввода пробы в инжектор и промывки инжектора; фильтрующее устройство с фильтром 0,45 мкм; ультразвуковая ванна для дегазации элюентов и подготовки пробы; общелабораторное оборудование (аналитические весы с точностью взвешивания до 0,1 мг; мерная стеклянная посуда).
Реактивы и растворы. Ацетонитрил градации для жидкостной хроматографии (210 - 230 нм), вода бидистиллированная или очистки супер-q на оборудовании "Миллипор". Подвижная фаза (элюент) - ацетонитрил:вода (80:20 по объему), дегазированная перед использованием с помощью ультразвуковой ванны или потока гелия. Сорбенты для прямофазной хроматографии типа сепарон SGX NH2, лихросорб NH2.
Выполнение анализа. Точную навеску технического продукта около 0,05 - 0,1 г (0,5 - 0,7 г жидкого концентрата) или аналитического стандарта определяемого вещества помещают в мерную колбу вместимостью 25 мл и растворяют в 5 - 7 мл хлороформа, затем проводят разбавление приготовленного раствора элюентом (из расчета получения концентрации в рабочей градуировочной смеси и испытуемом растворе 0,010 - 0,005 мг/мл) и 4 мкл приготовленного раствора вводят в хроматограф. Условия хроматографирования: колонка типа сепарон SGX NH2 5 мкм (150 x 3,3 мм); скорость элюента - 0,4 мл/мин; температура колонки 60 °C. Спектрофотометрические измерения проводят при длине волны 288 нм.
Из полученных хроматограмм рабочей градуировочной смеси определяют время удерживания и площадь хроматографического пика определяемого вещества в рабочей градуировочной смеси. По совладению времени удерживания на хроматограммах градуировочной смеси и хроматограммах анализируемой пробы проводят идентификацию веществ и вычисляют площади их хроматографических пиков в пробе.
Обработка результатов. Вычисление массовой доли ДВ в средстве X в процентах с применением абсолютной градуировки проводится по формуле:
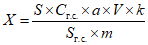 , где
, гдеS и Sг.с. - площади хроматографических пиков определяемого вещества в испытуемой пробе и рабочей градуировочной смеси;
Cг.с. - массовая концентрация аналитического стандарта определяемого вещества в рабочей градуировочной смеси, мг/мл;
a - массовая доля основного вещества в аналитическом стандарте, %;
V - объем раствора пробы, мл;
k - кратность разведения раствора пробы;
m - масса анализируемой пробы, мг.
В отсутствие стандартных образцов определяемых веществ при анализе дератизационных субстанций в виде малоочищенной технологической смеси (трифенацин, тетрафенацин) в качестве аналитического стандарта может быть принят стандарт дифенацина, имеющий квалификацию государственного стандартного образца. Массовую долю ДВ вычисляют по сумме площадей всех хроматографических пиков, характеризующихся спектрами индан-1,3-дионов.
При вычислении содержания примесей в техническом продукте может быть применен метод нормализации площадей.
Хроматографический метод определения индандионов с применением обращенно-фазной высокоэффективной хроматографии основан на применении УФ-детектирования, хроматографировании пробы в градиентном режиме и использовании абсолютной градуировки.
Хроматографирование проводят на колонках, заполненных сорбентом типа сепарон C18, ультрасфер ODS, партисил ODS 2; в качестве элюента используют смеси растворителей, приготавливаемые по объему: метанол:изопропанол:0,1%-й раствор фосфорной кислоты в соотношении 65:15:20, ацетонитрил:0,2%-й раствор фосфорной кислоты в соотношении 75:25, а также применяют другие хроматографические системы сорбент-элюент для обращенно-фазной хроматографии.
Метод избирателен. Спектрофотометрическому измерению дифенацина, этилфенацина, изопропилфенацина и хлорфасинона не мешают технологические и функциональные добавки, типичные для рецептурного состава приманок: битрекс, бензоат натрия, красители. В зависимости от состава пищевой основы экспериментально подбирают условия экстракционного выделения ДВ и очистки экстракта с помощью тест-пробы.
Выполнение анализа.
Подготовка пробы. В навеску зерновой приманки массой 10 - 20 г добавляют 100 мл органического растворителя и проводят экстрагирование с помощью ультразвуковой ванны в течение 30 - 50 мин. В качестве экстрагента может быть применен ацетонитрил; ацетонитрил, подкисленный уксусной кислотой (1% об.); ацетонитрил:метанол в соотношении 1:1 и др. После отстаивания отбирают аликвоту экстракта, фильтруют через фильтр 0,45 мкм и хроматографируют в градиентном режиме обращенно-фазной хроматографии.
При анализе зерновой приманки в виде парафинированного брикета может быть использована последовательная экстракция растворителями разной полярности. Сначала проводят экстракцию небольшими порциями гексана при интенсивном перемешивании на магнитной мешалке. Экстракцию повторяют несколько раз, проэкстрагированный гексаном образец подсушивают до воздушно-сухого состояния, после чего проводят экстракцию ацетонитрилом с помощью ультразвуковой ванны в течение 30 мин. В гексановый экстракт добавляют сорбент - оксид алюминия и после выдерживания в течение 20 - 30 мин фильтруют. Осадок на фильтре промывают ацетонитрилом, полученный фильтрат вносят в подсушенный образец и проводят экстракцию ацетонитрилом с помощью ультразвуковой ванны в течение 30 мин. После отстаивания отбирают аликвоту экстракта, фильтруют через фильтр 0,45 мкм и хроматографируют в градиентном режиме обращенно-фазной хроматографии.
Условия хроматографирования: колонка типа LUNA C18 3 мкм (150 x 3,3 мм); элюент А - ацетонитрил; элюент Б - 0,16%-й раствор фосфорной кислоты. Градиент по ацетонитрилу от 60 до 85% за 10 мин; скорость подвижной фазы 0,4 мл/мин; температура колонки комнатная; объем хроматографируемой дозы 10 мкл. Примерное время удерживания хлорфасинона 8,9 мин, дифенацина 10,2 мин.
Идентификацию определяемого вещества проводят сравнением времени удерживания в градуировочной смеси и анализируемой пробе при одной длине волны в интервале 250 - 280 нм, дополнительно могут быть использованы спектры поглощения в области 190 - 400 нм в исследуемой пробе и градуировочной смеси.
Обработка результатов. Массовую долю ДВ в приманке X в процентах с применением абсолютной градуировки вычисляют с учетом фактора извлечения  по формуле:
по формуле:
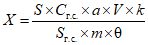 , где
, гдеS и Sг.с. - площади хроматографических пиков определяемого вещества в испытуемой пробе и рабочей градуировочной смеси;
Cг.с. - массовая концентрация аналитического стандарта определяемого вещества в рабочей градуировочной смеси, мг/мл;
a - массовая доля основного вещества в аналитическом стандарте, %;
V - объем раствора пробы, мл;
k - кратность разведения раствора пробы;
m - масса анализируемой пробы, мг;
зоокумарина и бромадиолона
Определение зоокумарина и бромадиолона в дератизационных средствах спектрофотометрическим методом может быть проведено по собственному поглощению в ультрафиолетовой части спектра и по измерению оптической плотности растворов натриевой соли.
Спектрофотометрический метод определения зоокумарина и бромадиолона по собственному поглощению в ультрафиолетовой части спектра основан на измерении оптической плотности раствора пробы в органическом растворителе с использованием молярного коэффициента поглощения определяемого вещества.
Приборы, реактивы и растворы. Спектрофотометр СФ-26 или другого типа; весы аналитические; ротационный испаритель; стеклянная мерная посуда; 2%-й раствор натрия гидроокиси; хлороформ; диоксан; этанол; оксид алюминия для хроматографии.
Выполнение анализа. Зоокумарин (3%) из навески порошкового средства массой около 0,1 г экстрагируют 25 мл диоксана с помощью магнитной мешалки и разбавляют диоксаном в 10 раз, после фильтрования проводят измерение оптической плотности раствора при длине волны 310 нм.
Обработка результатов. Содержание зоокумарина в средстве X в процентах вычисляют по формуле:
 , где
, гдеD и D0 - оптические плотности анализируемого раствора и раствора "холостой" пробы средства, не содержащей зоокумарина;
308,2 - молекулярная масса зоокумарина, г/моль;
V - объем фотометрируемого раствора, мл;
10 - кратность разведения экстракта;
m - масса средства, г;
l - толщина поглощающего слоя, см.
При определении бромадиолона (0,25%) в концентрате навеску средства массой около 0,04 г растворяют в 5 мл этанола. Измерения проводят при длинах волн 320 и 340 км. При вычислении содержания бромадиолона применяют разность молярных коэффициентов поглощения бромадиолона при 320 и 340 нм, равную 7800 л/моль·см.
Метод не избирателен в присутствии растворимых веществ рецептурного состава средства, поглощающих в указанных областях спектра. Метод может быть применен для серийных анализов в условиях производства.
Спектрофотометрический метод определения зоокумарина по поглощению его натриевой соли основан на измерении при 306 - 308 нм оптической плотности раствора натриевой соли зоокумарина, получаемой с помощью 2%-го раствора гидроокиси натрия.
Метод не избирателен в присутствии веществ в пробе, поглощающих при длине волны измерения 306 - 308 нм. Метод может быть применен для серийных анализов в условиях производства.
Выполнение анализа. При анализе технического продукта пробу растворяют в 2%-м растворе гидроокиси натрия и измеряют оптическую плотность при 306 - 308 нм.
При анализе препаративных форм проводят извлечение хлороформом, при необходимости - сорбционную очистку экстракта на колонке с окисью алюминия. Растворитель отгоняют и растворяют сухой остаток в 2%-м растворе гидроокиси натрия. После фильтрования измеряют оптическую плотность при 308 нм, используя в качестве раствора сравнения "холостую пробу". Количество ДВ измеряют по калибровочному графику в интервале 25, 50, 100 и 200 мкг, для построения которого применяют стандартный раствор натриевой соли зоокумарина 0,1 мг/мл.
Обработка результатов. Массовую долю зоокумарина в средстве X в процентах вычисляют по формуле:
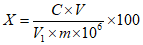 , где
, гдеC - содержание зоокумарина, найденное по калибровочному графику, мкг;
V - объем раствора средства, мл;
V1 - объем раствора, взятый для анализа, мл;
m - масса средства, взятая для анализа, г;
106 - коэффициент пересчета мкг в г.
антикоагулянтов оксикумаринового ряда
Определение антикоагулянтов кумаринового ряда (бродифакума, бромадиолона, дифетиалона, зоокумарина, куматетралила, флокумафена) в субстанциях (технических продуктах, жидких и порошковых концентратах) и различных препаративных формах дератизационных средств проводят методом прямой или обращенно-фазной высокоэффективной хроматографии с применением спектрофотометрического детектирования. Для идентификации и хроматографических измерений антикоагулянтов оксикумаринового ряда применяют спектрофотометрические детекторы для измерений при одной длине волны, а также детекторы на диодной матрице для измерений в области 190 - 400 нм, которые позволяют повысить надежность идентификации веществ за счет использования двух критериев: 1) спектр поглощения в области 190 - 400 нм; 2) время удерживания в заданных условиях хроматографических измерений.
Специфические условия подготовки пробы и спектрофотометрического детектирования обуславливаются содержанием ДВ и составом ингредиентов рецептуры средства (технологические и функциональные добавки, в т.ч. натуральные пищевые аттрактанты).
Субстанции (технические продукты и жидкие концентраты) растворяют в растворителе, совместимом с хроматографической подвижной фазой. Определение ДВ проводят в условиях прямой или обращенно-фазной высокоэффективной хроматографии. При анализе технического продукта определяют содержание основного вещества с использованием абсолютной градуировки или "внутреннего эталона", для количественной оценки примесей применяют метод нормализации площадей.
Из порошковых концентратов требуется выделение ДВ в органическую фазу жидкостной экстракцией.
Из пищевых приманок также извлекают ДВ с помощью жидкостной экстракции. При этом из-за низкого уровня содержания ДВ (3 - 10%) в пищевых приманках на фоне сложного рецептурного состава и пищевой основы повышаются требования к селективному выделению ДВ и условиям эффективного хроматографического разделения компонентов пробы.
Метод избирателен. Определению бродифакума, бромадиолона, дифетиалона, зоокумарина, куматетралила, флокумафена не мешают технологические примеси в техническом продукте.
Приборы. Аналитический жидкостный хроматограф "Стайер" или другого типа, снабженный спектрофотометрическим детектором на диодной матрице или детектором для измерений при одной длине волны в ультрафиолетовой области спектра, термостатируемой колонкой, изократической системой микронасосов, инжектором типа "Реодайн" (или другого типа) с дозирующей петлей объемом от 5 до 20 мкл, программой управления оборудованием и обработки хроматографических данных на базе персонального компьютера.
Хроматографическая колонка, как правило, заводского заполнения сорбентом, с соответствующей предколонкой. Микрошприц вместимостью 100 - 250 мкл для ручного ввода пробы в инжектор и промывки инжектора. Фильтрующее устройство с фильтром 0,45 мкм. Ультразвуковая ванна для дегазации элюентов и подготовки пробы. Общелабораторное оборудование (аналитические весы с точностью взвешивания до 0,1 мг, мерная стеклянная посуда).
Реактивы и растворы. Ацетонитрил градации для жидкостной хроматографии (210 - 230 нм); уксусная кислота х.ч.; вода бидистиллированная или очистки супер-q на оборудовании "Миллипор". Сорбенты для прямофазной и обращенно-фазной хроматографии типа сепарон SGX NH2, лихросорб NH2, MAX RP C12, зорбакс ODS, лихросорб RP 18 и др.
Элюент (подвижная фаза) - ацетонитрил:вода в соотношении 80:20 (по объему); ацетонитрил:вода:уксусная кислота в соотношении 80:20:1 (по объему), дегазированные перед использованием с помощью ультразвуковой ванны или потока гелия.
Выполнение анализа. Точную навеску технического продукта 0,05 г или аналитического стандарта определяемого вещества растворяют в мерной колбе вместимостью 50 - 100 мл в соответствующем органическом растворителе (ацетонитрил, этиловый спирт и др.) с помощью ультразвуковой ванны, затем проводят последовательное разбавление исходного раствора ацетонитрилом или элюентом (из расчета получения концентрации в рабочей градуировочной смеси и испытуемом растворе 0,010 - 0,005 мг/мл) и хроматографируют в условиях прямофазной или обращенно-фазной высокоэффективной хроматографии. Из полученных хроматограмм рабочей градуировочной смеси определяют время удерживания и площадь хроматографического пика определяемого вещества в рабочей градуировочной смеси. Из хроматограмм испытуемой пробы проводят идентификацию определяемого вещества по совпадению времени удерживания и вычисляют площадь хроматографического пика в испытуемой пробе. Применение диодно-матричного детектора позволяет записать и сравнить спектры поглощения хроматографического пика определяемого вещества в диапазоне 190 - 400 нм в рабочей градуировочной смеси и испытуемой пробе. Близким характером спектров поглощения характеризуются зоокумарин и куматетралил, бродифакум и бромадиолон.
Условия хроматографирования при прямофазной и обращенно-фазной высокоэффективной хроматографии приведены в табл. 4.7.
Таблица 4.7
Параметры | Прямофазная высокоэффективная хроматография | Обращенно-фазная высокоэффективная хроматография |
Подвижная фаза | Ацетонитрил:вода 80:20 | Ацетонитрил:вода:уксусная кислота 80:20:1 |
Скорость подвижной фазы | 0,5 мл/мин | 0,5 мл/мин |
Колонка | Сепарон NH2 5 мкм (150 x 3,3 мм) | Max RP (C12) 4 мкм (250 x 4 мм) |
Температура колонки | 37 °C | 37 °C |
Хроматографируемая доза | 20 мкл | 20 мкл |
В указанных условиях бродифакум и бромадиолон детектируются двумя пиками стереоизомеров. Примерное относительное время удерживания действующих веществ по основному пику бромадиолона приведено в табл. 4.8.
Таблица 4.8
Действующее вещество | Прямофазная высокоэффективная хроматография | Обращенно-фазная высокоэффективная хроматография |
Дифетиалон | 1,5 | 0,7 |
Бродифакум | 1,4 | 0,8 |
Бромадиолон | 1,0 | 1,0 |
Куматетралил | 0,9 | 1,8 |
Зоокумарин | 0,8 | 2,9 |
Обработка результатов. Вычисление массовой доли основного вещества X в процентах в техническом продукте с применением абсолютной градуировки проводится по формуле:
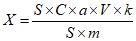 , где
, гдеS и Sр.г.с. - площадь хроматографического пика определяемого вещества в анализируемой пробе и рабочей градуировочной смеси;
C - массовая концентрация анализируемого стандарта определяемого вещества в рабочей градуировочной смеси, мг/мл;.
a - массовая доля действующего вещества в аналитическом стандарте, %;
V - объем раствора пробы, мл;
k - кратность разведения пробы;
m - масса средства, мг.
Определение ДВ в порошковых и жидких концентратах основано на разбавлении или экстракционном извлечении ДВ, разделении компонентов пробы в хроматографической системе сорбент - подвижная жидкая фаза (элюент) в изократическом режиме и последующем спектрофотометрическом измерении в элюате определяемого вещества с применением абсолютной градуировки.
Метод избирателен. Спектрофотометрическому измерению бродифакума, бромадиолона, дифетиалона, зоокумарина, куматетралила, флокумафена не мешают технологические и функциональные добавки, типичные для рецептурного состава концентратов: битрекс, бензоат натрия, красители (например, метиленовый голубой, красный темный) и растворители (например, этиловый и пропиловый спирт, триэтиленгликоль, пропиленгликоль).
Выполнение анализа.
Подготовка пробы. Масляный концентрат (около 0,1 г) растворяют в ацетонитриле в мерной колбе вместимостью 25 мл, затем 20 мл полученного раствора вносят в мерную колбу вместимостью 25 мл и добавляют до метки элюент (смесь ацетонитрил:вода в соотношении 80:20), после перемешивания хроматографируют.
Гелеобразный жидкий концентрат (около 0,5 г) помещают в мерную колбу вместимостью 25 мл и растворяют в 2,5 мл диметилформамида при периодическом перемешивании или с помощью ультразвуковой ванны, затем добавляют ацетонитрил. После отстаивания 2 мл прозрачного раствора вносят в мерную колбу вместимостью 25 мл и добавляют до метки элюент (смесь ацетонитрил:вода в соотношении 80:20), после перемешивания хроматографируют.
Порошковый концентрат (0,05 - 0,15 г) экстрагируют 50 мл ацетонитрила с помощью ультразвуковой ванны в течение 20 - 25 мин, затем центрифугируют и полученный раствор хроматографируют. В качестве экстрагента может быть применен также водный ацетонитрил (1:1), хлористый метилен, насыщенный муравьиной кислотой - в этом случае отгоняют экстрагент с помощью ротационного испарителя, сухой остаток растворяют в элюенте (метанол:вода:уксусная кислота в соотношении 94,2:5:0,8) и хроматографируют. При необходимости проводят сорбционную очистку экстракта от красителя на слое целита 545.
Условия хроматографирования при прямофазной и обращенно-фазной высокоэффективной жидкостной хроматографии. Для прямофазной высокоэффективной жидкостной хроматографии может быть использована хроматографическая система: аминная колонка с сорбентом типа сепарон SGX NH2 5 мкм, лихросорб NH2 5 мкм - элюент (ацетонитрил:вода в соотношении 80:20). Для обращенно-фазной высокоэффективной жидкостной хроматографии - хроматографическая система: колонка с сорбентом типа MAX RP C12, зорбакс ODS, нуклеосил C18, лихросорб RP 18 - (элюент ацетонитрил:вода:уксусная кислота в соотношении 80:20:1; метанол:вода:уксусная кислота в соотношении 94,2:5:0,8 и др).
Длина волны измерений может быть применена в диапазоне от 254 до 288 нм. Скорость элюента применяется в интервале от 0,4 до 2 мл/мин, температура колонки в диапазоне 25 - 60 °C, объем хроматографируемой пробы 5 - 20 мкл.
Рабочие градуировочные смеси готовят с массовой концентрацией определяемых ДВ 0,005 - 0,010 мг/мл. Из полученных хроматограмм рабочей градуировочной смеси определяют время удерживания и площадь хроматографического пика вещества в рабочей градуировочной смеси. Из хроматограмм испытуемой пробы проводят идентификацию определяемого вещества по совпадению времени удерживания и вычисляют площадь хроматографического пика в анализируемой пробе. Для идентификации определяемого вещества дополнительно могут быть использованы спектры поглощения в области 190 - 400 нм в исследуемой пробе и градуировочной смеси.
Обработка результатов. Массовую долю ДВ в концентрате определяют по известной формуле при применении абсолютной градуировки.
Определение ДВ в дератизационных приманках основано на его экстракционном извлечении, хроматографическом разделении компонентов пробы в градиентном режиме и последующем спектрофотометрическом измерении определяемого вещества в элюате с применением абсолютной градуировки.
Метод избирателен. Спектрофотометрическому измерению бродифакума, бромадиолона, дифетиалона, зоокумарина, куматетралила, флокумафена не мешают технологические и функциональные добавки, типичные для рецептурного состава приманок: красители (например, метиленовый голубой, красный темный), битрекс, бензоат натрия и растворители (например, этиловый и пропиловый спирт, триэтиленгликоль, пропиленгликоль). Идентификацию определяемого вещества проводят сравнением времени удерживания при одной длине волны в интервале 250 - 280 нм, дополнительно могут быть использованы спектры поглощения в области 190 - 400 нм в исследуемой пробе и градуировочной смеси.
Выполнение анализа. Зерновые приманки. Навеску зерновой приманки массой 10 - 20 г экстрагируют ацетонитрилом и обрабатывают в ультразвуковой ванне в течение 30 мин. В качестве экстрагента может быть применен ацетонитрил, подкисленный уксусной кислотой (1% об.). После отстаивания отбирают аликвоту экстракта, фильтруют через фильтр 0,45 мкм и хроматографируют в градиентном режиме обращенно-фазной хроматографии. Условия хроматографирования: колонка с сорбентом нуклеосил С18 (125 x 3 мм); элюент А - ацетонитрил, элюент Б (0,16% об.) - раствор ортофосфорной кислоты. Градиент по ацетонитрилу от 40 до 75% за 10 мин; 75% изократика 5 мин; скорость подвижной фазы 0,4 мл/мин; длина волны 284 нм; объем хроматографируемой дозы 10 мкл.
Гранулированные приманки. Навеску 20 - 40 г измельченной гранулированной приманки помещают в коническую колбу вместимостью 500 мл, добавляют 250 мл экстрагента и интенсивно перемешивают на магнитной мешалке в течение 20 мин. В качестве экстрагента применяют хлористый метилен, насыщенный муравьиной кислотой. Затем содержимое колбы количественно переносят на фильтр с целитом и трижды промывают экстрагентом порциями по 50 мл. Фильтрат упаривают с помощью ротационного испарителя. К сухому остатку добавляют 10 мл смеси метанол:хлористый метилен (3:2) (по объему), центрифугируют и раствор вводят в хроматограф. Условия хроматографирования: колонка с сорбентом для обращенно-фазной хроматографии зорбакс ODS 5 мкн (250 x 4,6 мм); подвижная фаза метанол:вода:уксусная кислота (94,2:5:0,8); скорость 1 мл/мин; длина волны 254 нм; объем хроматографируемой дозы 10 мкл. Рабочая градуировочная смесь в растворителе - метанол:хлористый метилен (3:2) с концентрацией определяемого вещества 0,2 мг/мл.
Обработка результатов. Массовую долю ДВ в приманке X в процентах с применением абсолютной градуировки вычисляют по следующей формуле с учетом фактора извлечения  :
:
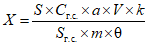 , где
, гдеS и Sг.с. - площади хроматографических пиков определяемого вещества в испытуемой пробе и рабочей градуировочной смеси;
Cг.с. - массовая концентрация анализируемого стандарта определяемого вещества в рабочей градуировочной смеси, мг/мл;
a - массовая доля основного вещества в аналитическом стандарте, %;
V - объем раствора пробы, мл;
k - кратность разведения раствора пробы;
m - масса пробы, взятая на анализ, мг;
1. Крейнгольд С.У. Практическое руководство по химическому анализу дезинфекционных препаратов. М., 2002. 156 с.
2. Шамб У., Сеттерфилд Ч., Вентворс Р. Перекись водорода. М., 1958. С. 460 - 468.
3. Губен-Вейль. Методы органической химии. Методы анализа. М.: Химия, 1967. С. 452 - 462.
4. Сукиасян А.Н., Копылова А.И. Количественное определение глутарового альдегида в разбавленных водных растворах//Теория и практика дезинфекции и стерилизации: Сборник научных трудов. М., 1983. С. 116 - 119.
5. Сукиасян А.Н., Свитова И.Р. Количественное определение надкислот в растворах, содержащих перекись водорода//Хим.-фарм. журнал. 1983. N 3. С. 366 - 368.
6. Сукиасян А.Н., Свитова И.Р. Модификация цериметрически-йодометрического метода анализа перекисных компонентов в растворах, содержащих надкислоту и перекись водорода//Проблемы дезинфекции и стерилизации: Сб. научн. тр. М., 1980. С. 115 - 119.
7. Газоанализатор 3.02П-Р. Руководство по эксплуатации ИРМБ. 4 133 12.005-02 РЭ. СПб, 2001.
8. Оптический анализатор озона "Циклон-5.51". Руководство по эксплуатации ИРМБ. 4 133 13.009 РЭ. СПб, 2001.
9. Мельников Н.Н., Новожилов К.Б., Белан С.Р. Пестициды и регуляторы роста растений: Справочник. М., 1995.
10. Методы определения микроколичеств пестицидов в продуктах питания, кормах и внешней среде. М.: Колос, 1992. Т. 1, 2. N 94.
11. Гигиенические критерии состояния окружающей среды, N 94, Перметрин, ВОЗ, Женева, 1992.
12. Гигиенические критерии состояния окружающей среды, N 82, Циперметрин, ВОЗ, Женева, 1991.
13. Гигиенические критерии состояния окружающей среды, N 97, Дельтаметрин, ВОЗ, Женева, 1992.
14. Гигиенические критерии состояния окружающей среды, N 87, Аллетрины, ВОЗ, Женева, 1992.
15. Гигиенические критерии состояния окружающей среды, N 96, d-Фенотрин, ВОЗ, Женева, 1993.
16. Крейнгольд С.У. и Шестаков К.А. Методика определения фосфида цинка в техническом препарате//Дезинфекционное дело, N 2. 2003. С. 23 - 24.
17. Хубер Л. Применение диодно-матричного детектирования в ВЭЖХ. М.: Мир, 1993. 95 с.
18. Методические указания по определению зоокумарина в тканях и крови животных, в приманках и препарате пенокумарин хроматографическими и спектрофотометрическими методами от 20.12.1976 N 1550-76//Методы определения микроколичеств пестицидов в продуктах питания, кормах и внешней среде. Справочное. Мин. сельхоз. СССР. Гос. Комиссия по химическим средствам борьбы с вредителями, болезнями растений и сорняками/Под ред. М.А. Клисенко. М.: Колос, 1983. С. 227 - 331.
19. Крейнгольд С.У. Простые и экспрессные методы химического контроля качества дезинфекционных препаратов//Дез. дело. N 4. 2002. С. 37 - 39.
20. Крейнгольд С.У. Спектрофотометрическое определение фенолов в дезинфицирующих средствах после разделения их методом тонкослойной хроматографии//Дез. дело, N 4. 2003. С 45 - 47.
21. Крейнгольд С.У. и Шестаков К.А. Определение триклозана в жидком туалетном антибактериальном мыле//Дез. дело, N 3. 2003. С. 46 - 47.
22. Мыло жидкое с дезинфицирующим эффектом "НИКА-СВЕЖЕСТЬ антибактериальное". ТУ 9392-023-12910434-2005. ООО НПФ "ГЕНИКС".
23. Средство дезинфицирующее "ДЕЛАСЕПТ-ГЕЛЬ" (кожный антисептик). Технические условия ТУ 9392-051-00479095-2005. ЗАО "Петроспирт".
24. Средство дезинфицирующее КЕМИ-САЙД. Технические условия ТУ 9392-001-5671Б5159-2004. ООО "КЕМИТРЕЙД".
эффективности дезинфицирующих и стерилизующих средств
Глава 5 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
оценки эффективности дезинсекционных средств
Глава 6 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
эффективности дератизационных средств
Глава 7 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
и опасности дезинфекционных средств
Глава 8 исключена. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.
КЛАССИФИКАЦИЯ ОПАСНОСТИ ВРЕДНЫХ ВЕЩЕСТВ
ПРИ КОЖНОМ ПУТИ ПОСТУПЛЕНИЯ
Приложение 1 исключено. - Р 4.2.3676-20, утв. Роспотребнадзором 18.12.2020.