СПРАВКА
Источник публикации
М.: ФГБУ "Институт стандартизации", 2025
Примечание к документу
Документ вводится в действие с 01.03.2026.
Название документа
"ГОСТ Р ИСО 20399-2025. Национальный стандарт Российской Федерации. Биотехнология. Средства лекарственные биологические для медицинского применения. Вспомогательные материалы, используемые при производстве клеточных препаратов"
(утв. и введен в действие Приказом Росстандарта от 24.10.2025 N 1276-ст)
"ГОСТ Р ИСО 20399-2025. Национальный стандарт Российской Федерации. Биотехнология. Средства лекарственные биологические для медицинского применения. Вспомогательные материалы, используемые при производстве клеточных препаратов"
(утв. и введен в действие Приказом Росстандарта от 24.10.2025 N 1276-ст)
Содержание
Приказом Федерального
агентства по техническому
регулированию и метрологии
от 24 октября 2025 г. N 1276-ст
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
БИОТЕХНОЛОГИЯ
СРЕДСТВА ЛЕКАРСТВЕННЫЕ БИОЛОГИЧЕСКИЕ
ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ
ВСПОМОГАТЕЛЬНЫЕ МАТЕРИАЛЫ, ИСПОЛЬЗУЕМЫЕ
ПРИ ПРОИЗВОДСТВЕ КЛЕТОЧНЫХ ПРЕПАРАТОВ
Biotechnology. Biological products for medical use.
Ancillary materials present during the production
of cellular therapeutic products
(ISO 20399:2022, Biotechnology - Ancillary materials present
during the production of cellular therapeutic products
and gene therapy products, IDT)
ГОСТ Р ИСО 20399-2025
ОКС 07.080
Дата введения
1 марта 2026 года
1 ПОДГОТОВЛЕН Федеральным государственным бюджетным учреждением "Российский институт стандартизации" (ФГБУ "Институт стандартизации") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 326 "Биотехнологии", Техническим комитетом ТК 458 "Разработка, производство и контроль качества лекарственных средств"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 24 октября 2025 г. N 1276-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 20399:2022 "Биотехнология. Вспомогательные материалы, присутствующие при производстве продуктов клеточной и генной терапии" (ISO 20399:2022 "Biotechnology - Ancillary materials present during the production of cellular therapeutic products and gene therapy products", IDT).
Наименование настоящего стандарта изменено относительно наименования указанного международного стандарта для приведения в соответствие с ГОСТ Р 1.5-2012 (пункт 3.5).
Международный стандарт разработан Техническим комитетом ИСО/ТК 276 "Биотехнология" Международной организации по стандартизации (ИСО).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты, сведения о которых приведены в дополнительном приложении ДА.
Дополнительные сноски в тексте стандарта, выделенные курсивом, приведены для пояснения текста оригинала
5 ВВЕДЕН ВПЕРВЫЕ
6 Некоторое элементы настоящего стандарта могут являться объектами патентных прав
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.rst.gov.ru)
Вспомогательные материалы (ВМ) - это материалы, которые вступают в контакт с клеточным препаратом во время манипуляций с клетками, но не предназначены для использования в составе готового препарата. Примеры ВМ приведены в приложении A.
ВМ могут иметь сложный состав из большого числа компонентов. К ВМ относят, например, соли, буферные растворы, питательные среды, добавки, такие как факторы роста, ферменты и антитела, применяемые для очистки продукта иммунологическими методами. Если ВМ включает несколько материалов, например питательные среды, то все его компоненты являются ВМ. Вариабельность их состава между сериями может оказывать негативное влияние на способность процесса производить препараты клеточной и генной терапии с требуемыми показателями качества.
Таким образом, ВМ могут повлиять на безопасность и эффективность препаратов клеточной и генной терапии. Приемлемый контроль ВМ определяется на основе риск-ориентированного подхода.
В настоящем стандарте установлены определения, связанные с ВМ.
Настоящий стандарт содержит рекомендации и требования к поставщикам и пользователям ВМ по обеспечению устойчивого производства и эффективному применению ВМ. В настоящем стандарте также приведена информация, которая может быть получена и предоставлена пользователям ВМ для демонстрации соответствия каждой партии ВМ в части идентичности, чистоты, хранения и стабильности, прослеживаемости, биобезопасности функциональных характеристик. Кроме того, в настоящем стандарте приведены рекомендации и требования, гарантирующие, что ВМ надлежащего качества позволит производить безопасные и эффективные готовые продукты.
В настоящее время ряд стандартов и руководящих документов устанавливают требования к надлежащему производству препаратов клеточной и генной терапии для обеспечения их безопасности и эффективности. Однако данные стандарты только косвенно относятся к ВМ. Настоящий стандарт не связан со стандартами, регулирующими требования по работе с клетками. В нем рассмотрены вопросы, связанные с ВМ, и обеспечивается прозрачность в отношении ожиданий поставщиков и потребителей ВМ.
Настоящий стандарт устанавливает требования и рекомендации в отношении поставщиков и пользователей вспомогательных материалов (ВМ) для улучшения стабильности и качества ВМ биологического (человеческого и животного) и химического происхождения, используемых при производстве препаратов клеточной и генной терапии для медицинского применения.
Настоящий стандарт распространяется на материалы, использующиеся в ходе производства клеток и вступающие в контакт с действующим веществом, но не вводятся преднамеренно в состав готового препарата клеточной и генной терапии для придания необходимых свойств.
Пример - Реактивы, антикоагулянты, цитокины, факторы роста, ферменты, антитела, сыворотки (человеческой или крупного рогатого скота), буферные растворы, питательные среды, чашки (покрытые биологическим материалом), гранулы (покрытые биологическим материалом), криопротекторы (средства для криоконсервации), активирующие агенты/реактивы, клетки, полученные не от млекопитающих (например, клетка насекомого, бактериальная клетка).
Настоящий стандарт не распространяется на материалы, которые не используют для манипуляций с клетками, не вступают в контакт с действующим веществом, или материалы, которые включены в состав готового препарата клеточной и генной терапии для придания необходимых свойств.
Пример - Клетки, которые являются исходными материалами, промежуточными продуктами или готовой формой препарата клеточной терапии, питающие клетки, добавки, используемые после окончания культивирования, каркасы, небиологические расходные материалы [например, стеклошарики (бусы), чашки, флаконы (матрасы) для культивирования, мешки, трубки, пипетки, иглы], другая пластиковая посуда, контактирующая с клетками или тканями, оборудование, инструменты.
Блок-схема принятия решений приведена в приложении A.
Примечание - Международные, национальные и/или региональные регламенты/требования также могут применяться к конкретным положениям, рассматриваемым в настоящем стандарте.
В настоящем стандарте использована нормативная ссылка на следующий стандарт [для датированных ссылок применяют только указанное издание ссылочного стандарта, для недатированных - последнее издание (включая все изменения)]:
ISO 8601-1, Date and time - Representations for information interchange - Part 1: Basic rules (Дата и время. Представление для обмена информацией. Часть 1. Основные правила)
В настоящем стандарте применены следующие термины с соответствующими определениями.
ИСО и МЭК поддерживают терминологические базы данных, используемые в целях стандартизации, по следующим адресам:
- платформа онлайн-просмотра ИСО: доступна по адресу https://www.iso.org/obp;
- Электропедия МЭК: доступна по адресу https://www.electropedia.org/.
3.1 действующее вещество (active substance): Вещество, обладающее требуемой биологической активностью в составе клеточного препарата (3.9) для его заявленного применения.
3.2 вспомогательный материал (исходное сырье) <*>; ВМ (ancillary material, raw material, AM): Материал, контактирующий с клеточным препаратом (3.9) в ходе технологического процесса, за исключением каркаса, небиологического расходного материала и пластиковой посуды, но не предназначенный для придания лекарственной форме необходимых свойств.
Примечание 1 - ВМ может быть критически важным для качества и безопасности клеточного препарата вследствие контакта в ходе технологического процесса.
Примечание 2 - Блок-схема принятия решения, которая указывает, относится ли материал к области применения настоящего стандарта, приведена в приложении A.
--------------------------------
<*> В соответствии с законодательством Российской Федерации и ЕАЭС ВМ относятся к исходному сырью и не являются вспомогательным веществом (ВВ) и исходным материалом.
3.3 примесь вспомогательного материала; примесь ВМ (ancillary material impurity, AM impurity): Любой компонент, присутствующий в ВМ (3.2), который является нежелательным.
3.4 поставщик вспомогательного материала; поставщик ВМ (ancillary material supplier, AM supplier): Организация, производящая и/или поставляющая ВМ (3.2) пользователю ВМ (3.5).
3.5 пользователь вспомогательного материала; пользователь ВМ (ancillary material user, AM user): Организация, использующая ВМ (3.2) в ходе технологического процесса производства клеточного препарата (3.9).
3.6 отсутствие компонентов животного происхождения; ADCF (animal-derived component free, ADCF): Характеристика, указывающая на отсутствие материала(ов) человеческого или животного происхождения.
Примечание 1 - Основной целью определения типов ADCF является обеспечение пользователя ВМ (3.2) информацией, необходимой для оценки риска (3.13).
Примечание 2 - В некоторых случаях вместо сокращения ADCF используют сокращение AOF (animal origin free), означающее "без компонентов животного происхождения".
Примечание 3 - В случае отсутствия компонентов человеческого и животного происхождения, как правило, применяют термин "не содержащий ксеногенов".
3.7 биологический материал (biological material): Любое вещество, происходящее от органического объекта, или его полученная часть, такого как человек, животное, растение, микроорганизм(ы) или многоклеточный(ые) организм(ы), не относящий(е)ся к животным и растениям (например, бурые водоросли, грибы).
[ИСО 20387:2018, 3.7]
3.8 биобезопасность (biosafety): Процедуры и средства контроля, снижающие риск непреднамеренного воздействия или высвобождения биологических материалов (3.7).
Примечание - Данное определение включает непреднамеренное воздействие, например со стороны патогенов и токсинов, или их случайное высвобождение как риск для биобезопасности.
[ИСО 35001:2019, 3.22, изменено - добавлено примечание]
3.9 клеточный препарат <*> (cellular therapeutic product): Препарат, содержащий клетки в качестве действующего вещества (3.1) и используемый для целей клеточной или генной терапии.
--------------------------------
<*> Термину "клеточный препарат", используемому в настоящем стандарте, соответствует термин "высокотехнологический лекарственный препарат (ВТЛП)", установленный пунктом 6.3 статьи 4 Федерального закона от 12 апреля 2010 г. N 61-ФЗ "Об обращении лекарственных средств".
К ВТЛП относятся генотерапевтические лекарственные препараты (пункт 7.2 статьи 4 ФЗ N 61 от 12 апреля 2010 г.), лекарственные препараты на основе соматических клеток (препараты для терапии соматическими клетками) (пункт 17.2.2 Решения Совета ЕЭК от 3 ноября 2016 г. N 78), тканеинженерные лекарственные препараты (пункт 17.2.3 Решения Совета ЕЭК от 3 ноября 2016 г. N 78).
Пример - Препараты для клеточной и генной терапии, препараты тканевой инженерии, лекарственные препараты.
Примечание 1 - Препараты, полученные из клеток для генной терапии, включены в определение клеточного препарата, так как клетки не всегда являются действующим веществом для всех видов генной терапии.
Примечание 2 - Рекомбинантные белки не включены в данное определение клеточного препарата.
3.10 непрерывная ответственность в цепи поставок (chain of custody): Ответственность за материалы или контроль материалов на каждом этапе процесса производства.
Примечание - Цепь поставок - это непрерывный путь поставки ВМ (3.2) от начала производства до момента получения конечным пользователем ВМ (3.5). Она включает контроль, распределение и логистику до пользователя ВМ.
3.11 химически охарактеризованный компонент (chemically defined component): Вещество, химическая молекулярная структура которого идентифицирована/известна.
3.12 сертификат анализа; CoA (certificate of analysis, CoA): Документ, подтверждающий, что ВМ (3.2) прошел установленные испытания с указанными в нем результатами.
Примечание 1 - CoA, как правило, включает фактические результаты испытаний, выполненных при контроле качества отдельной серии ВМ.
Примечание 2 - Как правило, CoA отражает соглашение между поставщиком ВМ (3.4) и пользователем ВМ (3.5) о качестве ВМ.
[Руководство ИСО 73:2009, 3.4.1]
3.14 менеджмент риска (risk management): Скоординированные действия по руководству и управлению организацией в отношении рисков (для качества).
[Руководство ИСО 73:2009, 2.1]
3.15 срок годности (shelf life): Период, в течение которого, как ожидается, ВМ (3.2) будет соответствовать спецификациям (3.16) при хранении в установленных условиях.
3.16 спецификация (specification): Перечень испытаний, ссылок на аналитические методики и соответствующие критерии приемлемости, которые должны быть выполнены для подтверждения пригодности к предполагаемому применению.
3.17 стабильность (stability): Показатель, характеризующий способность материала, при хранении в установленных условиях, поддерживать значение(я) заявленного(ых) параметра(ов) в заданных пределах в течение заявленного периода времени.
[Руководство ИСО 30:2015, 2.1.15, изменено - заменено "стандартный материал" на "материал" и "значение установленного параметра" на "значение(я) заявленного(ых) параметра(ов)". Исключено примечание 1]
3.18 прослеживаемость (traceability): Способность отследить историю, применение или местонахождение объекта.
Примечание 1 - При рассмотрении продукта или услуги прослеживаемость может относиться:
- к происхождению материалов и составных частей;
- истории производства;
- распределению и местонахождению продукта или услуги после доставки.
Примечание 2 - В области метрологии принято определение, приведенное в Руководстве ИСО/МЭК 99.
[ИСО 9000:2015, 3.6.13]
3.19 спецификация требований пользователя; URS (user requirement specification, URS): Документ, в котором приведены спецификации (3.16) на ВМ (3.2), основанные на требованиях пользователя ВМ (3.5) к производству желаемого клеточного препарата (3.9).
ВМ - вспомогательный материал (ancillary material);
КК - контроль качества (quality control);
СМК - система менеджмента качества (quality management system);
ADCF - отсутствие компонентов животного происхождения (animal-derived component free);
AOF - без компонентов животного происхождения (animal origin free);
BSE - губчатая энцефалопатия крупного рогатого скота (bovine spongiform encephalopathy);
CoA - сертификат анализа (certificate of analysis);
CoC - сертификат соответствия (certificate of compliance);
CoI - сертификат радиационной обработки (certificate of irradiation);
CoO - сертификат происхождения (certificate of origin);
EDQM CEP - сертификат соответствия Европейского директората по качеству лекарственных средств и здравоохранению (European Directorate for the Quality of Medicines and Healthcare certificate of suitability);
GMP - надлежащая производственная практика (good manufacturing practice);
ICH - Международный совет по гармонизации технических требований к лекарственным средствам для медицинского применения (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use);
RP-HPLC - обратно-фазовая высокоэффективная жидкостная хроматография (reverse phase high performance liquid chromatography);
SDS - паспорт безопасности материала (safety data sheet);
SDS-PAGE - электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (sodium dodecyl sulfate poly acrylamide gel electrophoresis);
TSE - трансмиссивная губчатая энцефалопатия (transmissible spongiform encephalopathy);
USP - Фармакопея США (United States Pharmacopeia).
5.1 Основные положения о ВМ
Выбор ВМ для каждого клеточного препарата определяется технологическим процессом и готовой формой такого препарата (см. приложение A).
Пользователи ВМ несут ответственность за установление и поддержание статуса квалификации ВМ в процессах, включая соответствующий контроль ВМ. Объем контроля должен быть пропорционален рискам, связанным с данным ВМ, в отношении происхождения, производства или непрерывности цепочки поставок. Для выбора и квалификации ВМ следует применять риск-ориентированный подход.
ВМ могут влиять на показатели качества клеточного препарата:
a) качество и его постоянство необходимы для ВМ, которые были определены как критические для технологических процессов производства клеточных препаратов;
b) безопасность и непрерывная ответственность в цепи поставок критичны для ВМ, используемых в производстве клеточных препаратов.
Действия пользователя ВМ по оценке и контролю влияния ВМ на показатели качества клеточных препаратов основаны:
- на информации, предоставленной поставщиком ВМ;
- информации, полученной пользователем ВМ или поставщиком ВМ, либо обоими, в результате характеризации и тестирования ВМ или при производстве клеточного препарата;
- опубликованных стандартах или других научных методах, прошедших экспертную оценку (или эквивалентных им).
5.2 Типовые действия и ответственность сторон на разных этапах поставки ВМ
Блок-схема типового процесса поставки ВМ от поставщика пользователю приведена в приложении B.
Пользователь и поставщик могут согласовать спецификации на ВМ, предназначенные для клеточных препаратов, используя данную блок-схему.
Блок-схема типового процесса предназначена для определения зон ответственности пользователя(ей) и поставщика(ов) при использовании ВМ при производстве клеточного препарата.
В таблице 1 приведены рекомендации ответственным сторонам, выполняющим различные действия, и их ответственность.
Примечание - Необходимо, чтобы взаимоотношения между пользователем ВМ и поставщиком ВМ строились на сотрудничестве и прозрачности. Многие зоны ответственности представляют собой их совместные усилия. Данные виды деятельности получают выгоду от взаимоотношений между поставщиком и пользователем. Отсутствие таких отношений может привести к возникновению дополнительного риска для пользователя, например к отсутствию технической поддержки со стороны поставщика. Однако ответственность за эти действия определяется в каждом конкретном случае в индивидуальном порядке.
Таблица 1
ведущих деятельность по производству клеточных препаратов
Действие | Ответственная сторона | Ссылка на дополнительную информацию |
Предоставляет документированные доказательства того, что ВМ безопасен в отношении заболеваний животных, являющихся источником материала (например, BSE/TSE) | Поставщик ВМ | |
Готовит и подает мастер-файл на ВМ, если это применимо | Поставщик ВМ | |
Оценивает стабильность ВМ | Поставщик ВМ | |
Информирует пользователя ВМ о любых изменениях, которые с большой вероятностью или с уверенностью повлияют на ВМ (например, в соответствии с соглашением по качеству) | Поставщик ВМ | |
Проводит оценку системы "контейнер-укупорка" ВМ | Поставщик ВМ | |
Предоставляет CoA, CoO и SDS на ВМ | Поставщик ВМ | |
Проводят определение характеристик ВМ и разрабатывают файл спецификаций (идентификация, чистота, функциональные параметры, вирусная контаминация, животное происхождение) | Поставщик ВМ и пользователь ВМ | |
Выполняют соглашение по качеству и о поставках | Поставщик ВМ и пользователь ВМ | |
Предоставляет поставщику ВМ спецификации требований пользователя | Пользователь ВМ | |
Проводит риск-ориентированный процесс квалификации поставщиков ВМ, и, как правило, включающий первоначальный отбор, аудит производственной площадки, формализованное одобрение, постоянный мониторинг/надзор | Пользователь ВМ | |
Определяет, требуется ли исследование биосовместимости, биораспределения, цитотоксичности или тестирование на наличие вирусов (или, если применимо, есть возможность получить результаты тестирования у поставщика ВМ) | Пользователь ВМ | |
Проводит оценку рисков при использовании ВМ на основе информации, предоставленной поставщиком ВМ, или в сотрудничестве с поставщиком ВМ, например методом анализа видов и последствий отказов (FMEA) | Пользователь ВМ | |
Вводит аналогичные меры и планы для альтернативных поставщиков | Пользователь ВМ | |
Проводит квалификацию ВМ для заявленного применения | Пользователь ВМ | |
Подтверждает результаты испытания, приведенные в CoA, критического для клеточного препарата (например, функциональный анализ) | Пользователь ВМ | |
Оценивает влияние вариабельности серий ВМ на готовый клеточный препарат | Пользователь ВМ | |
Разрабатывает и внедряет план квалификации (стратегию контроля) ВМ для использования | Пользователь ВМ | |
Квалификация ВМ включает:
a) физико-химические характеристики ВМ, включая характеристики и показатели качества материала (например, идентификация, чистота, стабильность, эффективность и функциональные характеристики);
b) документацию по всем ВМ, включая их состав, качество или категорию, источник происхождения, каждого компонента, концентрацию (количество) и чистоту.
Примечание 1 - Состав и концентрация (количество) каждого компонента ВМ могут являться конфиденциальной информацией. Рекомендуется указывать ссылку на регистрационное досье на лекарственный препарат (DMF <*>).
Примечание 2 - В приложении E приведены примеры декларации качества произведенных биологических материалов, используемых в производстве клеточного препарата;
c) доказательства однородности серий ВМ между собой на предполагаемом этапе производства клеточного препарата, главным образом в отношении идентификации и функциональных характеристик ВМ;
d) приемлемый уровень биобезопасности, включая предотвращение контаминации посторонними вирусами, которые могут оказать негативный эффект на лекарственный препарат прямо или косвенно - на пациентов;
e) риск контаминации патогенными или токсическими биологическими и небиологическими агентами; соответствующий показатель, такой как предел обнаружения (LOD <*>) или предел переносимости (LOT <**>), должен быть определен и подтвержден, при необходимости;
--------------------------------
f) устойчивость и постоянство заявленного действия (функции) ВМ:
- ВМ должен выполнять заявленную функцию в типовом процессе производства клеточного препарата, выбранного поставщиком ВМ и соответствующего заявленному применению ВМ;
g) сопровождающую документацию:
- поставщик ВМ должен предоставить достаточный объем документов, содержащих информацию о ВМ, для того, чтобы пользователи ВМ могли обеспечить качество своих клеточных препаратов.
Примечание - В некоторых случаях объем документации ограничен в результате защиты интеллектуальной собственности;
h) декларации качества и снижения рисков при использовании ВМ для производимых биологических материалов, применяемых при производстве клеточных препаратов;
i) характеризация биологических материалов;
j) управление изменениями ВМ.
Поставщик ВМ отвечает за квалификацию ВМ в части их общих функциональных характеристик, а пользователь ВМ несет ответственность за квалификацию материалов в отношении предполагаемого применения.
Пользователь ВМ несет ответственность за квалификацию всех ВМ, используемых при производстве клеточных препаратов. Поставщики ВМ должны предоставить результаты испытаний на идентификацию и чистоту, если таковые имеются. При привлечении субподрядчиков у поставщиков ВМ должен быть план на случай, если субподрядчики не смогут провести квалификацию ВМ.
При необходимости, пользователь ВМ должен оценить наличие остатков ВМ в готовом клеточном препарате.
Пользователь ВМ должен провести аудит поставщика ВМ, чтобы обеспечить квалификацию материала.
5.4.1 Общие положения
Материалы биологического происхождения, в частности, человеческого или животного происхождения, могут представлять собой особый риск, включая перенос вирусов или контаминацию биологическими примесями, который не обязательно ограничивает использование компонентов биологического происхождения для производства ВМ или материалов, используемых в дальнейшем при производстве клеточных препаратов. Таким образом, при выборе и квалификации ВМ рекомендуется использовать риск-ориентированный подход.
Для обеспечения безопасности компонентов животного происхождения следует использовать риск-ориентированный подход.
Ключевые вопросы, которые должны быть рассмотрены, в частности в отношении происхождения материала (человеческого или животного происхождения):
a) Подвергается ли компонент финишной стерилизации?
b) Какое происхождение биологического(их) материала(ов)?
c) Прослеживается ли биологический(е) материал(ы) до источника происхождения?
d) Какие меры по снижению рисков применялись к биологическому(им) материалу(ам) кроме аудита поставщика(ов) ВМ?
Пример 1 - Получение от поставщика с низким уровнем риска TSE, этапы удаления или инактивации вируса, использование материалов фармацевтического качества, удаление вируса из ВМ, его компонента или подкомпонента или проверка на присутствие вирусов.
e) Существует ли риск контаминации ВМ вследствие контакта со смазывающими веществами (добавками, используемыми для уменьшения трения между пленками и между пленкой и оборудованием), а также в процессе производства?
Пример 2 - Недостаточно валидированные этапы удаления или инактивации вирусов, отсутствие проверки на наличие вирусов, отсутствие максимальной защиты от проникновения в производственные помещения насекомых и/или животных.
Поставщик должен осуществлять постоянные и надежные поставки ВМ пользователю. Пользователь ВМ должен установить аналогичные отношения с альтернативными поставщиками ВМ.
5.4.3 Инактивация вирусов
Если при производстве или в составе ВМ используют биологические материалы животного и/или человеческого происхождения, следует предусмотреть этап(ы) удаления или инактивации вирусов с учетом оценки риска воздействия ВМ на клеточный препарат. Инактивация может быть выполнена на уровне ВМ, на уровне компонентов или на комбинации этих уровней. Данные процессы должны быть валидированы, документация о валидации должна быть доступна пользователю ВМ.
Препараты крови человека, используемые в производстве ВМ, должны пройти процедуру удаления/инактивации вирусов.
Должно быть предоставлено документальное подтверждение проведения испытаний на наличие вирусов или применения мер по снижению риска вирусной контаминации при отсутствии процедуры инактивации вирусов, например проведение испытаний свиного трипсина на наличие соответствующих свиных вирусов или меры по снижению риска в случаях, когда субпоставщик не выполняет процедуры по удалению вирусов.
6.1 Критерии выбора ВМ
Пользователи ВМ должны учитывать ряд факторов при оценке пригодности биологического материала для производства клеточного препарата, независимо от класса (категории) или стандарта качества, заявленного поставщиком этого ВМ (см. таблицу 2).
Таблица 2
ключевые вопросы при оценке пригодности материалов
для производства клеточного препарата
Фактор | Основные требования и рекомендации | Основные вопросы |
Источник | Обеспечение качества ВМ (идентификации, активности, чистоты и других показателей качества) начинается с определения их источника или происхождения. Также существуют дополнительные риски, контаминации вирусами или биологическими примесями. Пользователь ВМ должен применять риск-ориентированный подход при выборе ВМ. Следует обратить внимание на самую последнюю информацию, включая, например, регуляторную и нормативную документацию, которая может служить приемлемым руководством по выбору подходящих источников происхождения биологических ВМ | Материал получен из источника, при котором риск контаминации вирусами снижен? Например, в [26] представлена актуальная информация о материале, получаемом от крупного рогатого скота в странах, где случаев TSE не обнаружено. Возможно заменить выбранный ВМ на другой с более низким уровнем риска? Например, заменить свиной трипсин на рекомбинантный трипсин с профилем вирусной безопасности. Имеет ли ВМ документированный профиль вирусной безопасности? |
Производство | Пользователям ВМ следует стремиться к получению максимального объема информации о процессе производства биологического ВМ, например ВМ может быть небиологического происхождения, но в процессе его производства может быть использован материал биологического происхождения | ВМ производится на выделенном производственном участке, например, чтобы свести к минимуму возможную перекрестную контаминацию? Если ВМ производят не на выделенном производственном участке, какие меры принимают для предупреждения контаминации при производстве других ВМ, например очистка линии, очистка производственного участка, оборудования, или при хранении других ВМ? Если ВМ не используют на выделенном производственном участке, какие меры предосторожности применяют для предотвращения перекрестной контаминации этого материала? Например, процедуры очистки линии, уборка помещений. Использовались ли биологические материалы при производстве ВМ? Если использовался рекомбинантный белок, он был произведен с использованием клеток животных или бактерий? Если ВМ был произведен с использованием компонентов животного происхождения или если материал получен из животного сырья, были ли проведены мероприятия по уменьшению количества потенциальных патогенов? Если да, были ли эти мероприятия валидированы? |
Испытания | Следует применять принципы, изложенные в руководстве ICH Q5A(R1), включая испытания на наличие вирусов в ВМ, проверку возможности технологического процесса удалять вирусы, а также испытание на вирусную контаминацию. Как правило, применяют радиационную или термическую обработку ВМ, например, питательных сред для культивирования клеток или сывороток. При этом следует обратить внимание, что она может привести к вариабельности серий. Должна быть установлена взаимосвязь между количественным содержанием или другими испытаниями и протоколами пользователя | Каким образом проводят характеризацию биологического ВМ, чтобы отразить его идентичность, чистоту и уровень эффективности? Если применимо, были ли проведены поставщиком ВМ надлежащие операции по вирусной инактивации/испытания на безопасность перед его выпуском в обращение? Имеется ли у ВМ документированный профиль вирусной безопасности? |
Прослеживаемость | Информация о прослеживаемости ВМ (от источника до поставщика) должна быть как можно более подробной, чтобы гарантировать, что любые последующие шаги в цепочке поставок не приведут к дополнительным рискам для безопасности или качества ВМ, например, в результате контаминации. Информация о прослеживаемости компонентов ВМ должна регистрироваться и быть доступна для аудита для гарантии вирусной безопасности ВМ | Ведет ли поставщик записи о ВМ и всех его компонентах, что обеспечивает контроль рисков, связанных с ВМ? В частности, возможно ли проследить происхождение этих ВМ, компонентов и подкомпонентов биологического происхождения до их источника? Внедрена ли на производстве ВМ СМК? Если да, то насколько она достаточна для обеспечения безопасности и применения ВМ? |
Бесперебойность поставок | После идентификации ВМ поставщик должен убедиться, что он может соответствовать требованиям, предъявляемым к постоянным и надежным поставкам этого ВМ. Любые сложности с поставками ВМ соответствующего качества, могут негативно сказаться на производстве клеточных препаратов. Пользователь ВМ должен оценить, в какой степени альтернативные поставщики ВМ могут рассматриваться как равноценные. Может потребоваться аттестация биологических функций, а также качества ВМ. В тех случаях, когда ВМ соответствующего качества отсутствует в продаже и не может пройти квалификацию надлежащим образом, он может быть изготовлен либо собственными силами, либо по контракту и под соответствующим контролем с эквивалентной прослеживаемостью. Однако пользователь ВМ должен сопоставить стабильность поставок со стоимостью и временем, затрачиваемым на полноценный КК ВМ, который может быть обеспечен при покупке у поставщика ВМ | Какие существуют альтернативы, если ВМ больше не доступен? Какие характеристики необходимо учесть для того, чтобы альтернативный ВМ можно было считать эквивалентным? Если подходящей альтернативы нет, можно ли изготовить ВМ собственными силами? Готов ли поставщик подписать контракт на поставку? Какого качества предлагаемый материал? См. также примеры признанных деклараций качества, используемых поставщиками для ВМ, используемых при производстве клеточных препаратов, приведенные в таблице E.1. Ограничен ли поставщик в выборе источников поставок? Указаны ли сроки выполнения заказа? Существуют ли особые требования к обращению с ВМ для обеспечения его функциональных характеристик? Существуют ли требования к мерам предосторожности для обеспечения стерильности процесса? |
--------------------------------
<*> Под термином "up-stream" понимают первый этап наработки биомассы в технологическом процессе производства.
6.2 Снижение риска
6.2.1 Научный подход
После поставки ВМ биологического происхождения поставщиком ответственность за обеспечение пригодности ВМ для использования по назначению лежит исключительно на пользователе ВМ. Таким образом, пользователь ВМ должен учитывать ряд факторов при использовании ВМ в процессе производства клеточных препаратов.
Таблица 3 содержит перечень факторов, которые должны приниматься во внимание пользователями ВМ для снижения рисков, связанных с использованием ВМ биологического происхождения, независимо от класса (категории) или стандарта качества, заявленных поставщиком ВМ.
Таблица 3
связанные со снижением рисков, возникающих при использовании
биологических материалов в производстве клеточных препаратов
Фактор | Основные принципы и рекомендации | Основные вопросы |
Верификация для конкретного применения или рассматриваемого процесса | Один ВМ может быть использован в нескольких отличных друг от друга и, как правило, сложных производственных процессах (например, сложные взаимосвязи между ферментами затрудняют экстраполяцию для другого применения соответствующего плана эксперимента) производства клеточных препаратов. Например, определенные цитокины и/или факторы роста присутствуют в разных процессах культивирования клеток и продуктах. Пользователю ВМ не следует предполагать, что ВМ, подходящий для одного этапа производственного процесса, подходит и для других этапов процесса. Таким образом, пользователь ВМ должен учитывать верификационные исследования, которые определяют влияние биологического ВМ на качество готового продукта применительно к конкретному технологическому процессу | Каковы конкретные риски и влияние на качество готового продукта клеточной или генной терапии, если ВМ не совместим с продуктом или процессом? На каких этапах технологического процесса используют ВМ и обособлен ли он в процессе производства от других компонентов? Какие методы стерилизации или другой деконтаминации были применены? Производят ли ВМ в условиях СМК, соответствующей определенному стандарту качества? Какой тип материала человеческого происхождения использован и из какой ткани он получен? В каком объеме было проведено обследование донора? В какой стране проживает донор? Соответствовали ли донация и поставка материала регуляторным требованиям, например к получению согласия донора? |
Испытания и показатели качества | Несмотря на то, что биологический ВМ не представляет прямого риска для безопасности, он все равно может быть непригодным для использования, если он не обеспечивает стабильно достаточный уровень биологической активности при его применении. Показатели качества в CoA, предоставленном поставщиком биологического ВМ, могут использоваться в качестве отправной точки, но они не должны быть единственной основой для обеспечения качества, так как CoA в большей степени содержит только базовую информацию, такую как стерильность и чистоту. Пользователь ВМ несет ответственность за оценку каждой партии поступающего материала. Аналитические методики испытаний, непосредственно влияющих на безопасность и качество готового продукта, подлежат валидации на начальных этапах. Необходимо продемонстрировать пригодность всех аналитических методик испытаний для предполагаемого применения | Какие показатели качества ВМ критичны для качества готового продукта? Как проводят контроль качества ВМ и подходят ли используемые аналитические методики или реактивы для этой цели? Учитывается ли потенциальная вариабельность серий ВМ в тех случаях, когда невозможно идентифицировать и изолировать одну партию ВМ, наиболее подходящую для производства клеточного препарата? По каким технологическим параметрам необходимо обеспечить однородность серий ВМ между собой? На основе чего проводится оценка продукта, например, по результатам испытаний для выпуска в обращение или спецификации? Содержатся ли в ВМ какие-либо компоненты животного происхождения, и если да, то какого типа? Какие испытания на наличие вирусов были проведены на ВМ и были ли предприняты какие-либо процедуры по удалению/инактивации вирусов? Если используется материал крупного рогатого скота, были ли предприняты какие-либо шаги для минимизации риска контаминации TSE |
Основным способом повышения уверенности пользователя в качестве ВМ является аудит поставщика. Аудит поставщика ВМ предоставляет пользователю ВМ возможность убедиться в наличии достаточности зарегистрированной и документированной информации, демонстрирующей прослеживаемость ВМ от его происхождения до конечного потребления. Если поставщик ВМ работает в сертифицированной СМК, это может дать дополнительные гарантии пользователю ВМ в том, что система хранения записей поддерживается в рабочем состоянии и существуют процедуры, обеспечивающие состояние управляемости процесса. Эти аудиты используются для оценки всего процесса производства, транспортирования и поставок, а также любых испытаний. Для каждого КК, проводимого поставщиком ВМ (например, внутрипроизводственного контроля, выпускного КК готового продукта), должна быть разработана стандартная операционная процедура, а также необходимо наличие соответствующей записи об обучении каждого сотрудника, проводящего данные испытания.
Пользователи ВМ должны проводить аудит поставщиков ВМ до начала поставок ВМ и впоследствии через определенные интервалы времени, чтобы гарантировать поддержание установленного уровня качества ВМ.
Перед проведением аудита поставщика ВМ пользователь ВМ может отправить поставщику анкету (опросник), содержащую ряд основных вопросов, которые позволят пользователю ВМ оценить пригодность и качество рассматриваемого ВМ.
Анкета поставщика ВМ может быть использована для получения информации о рассматриваемом ВМ, и должна быть достаточно полной, чтобы обеспечить оценку любых рисков для безопасности.
Биологические ВМ должны подвергаться дополнительной тщательной оценке в дополнение любых заявлений поставщика относительно качества ВМ. В таблицах 2 и 3 приведены примеры вопросов, которые пользователь ВМ может включить в анкету поставщика ВМ.
При использовании сыворотки крупного рогатого скота пользователь ВМ должен обеспечить выполнение специальных руководств по ее использованию при производстве клеточных препаратов.
Пользователь ВМ должен запросить CoA и CoO, которые подтверждают соответствие спецификации, состав и происхождение ВМ.
Пользователь ВМ может использовать информацию, предоставленную поставщиком биологического ВМ в качестве основы для оценки риска. Целью оценки риска в таких случаях является проведение оценки вероятности возникновения и тяжести вреда, связанного с конкретным риском конкретного ВМ путем использования систематической и последовательной процедуры.
Проведение оценки рисков может быть трудоемким мероприятием, и необходимо определить приоритетность наиболее высоких рисков. Пользователям следует использовать соответствующие инструменты оценки рисков (например, развертывание функции качества) для их количественной оценки (или для определения приоритетного числа рисков). Оценку рисков следует проводить для более широкой процедуры менеджмента рисков, включающей идентификацию, оценку (т.е. вероятность возникновения), контроль и мониторинг.
Пользователь ВМ должен определить приемлемый уровень риска и меры по снижению рисков. Необходимо обосновать используемые критерии для определения низкого, среднего и высокого уровня риска ВМ, а применяемая методология должна гарантировать, что уровень низкого риска достичь сложнее, чем уровень среднего или высокого риска. Также степень снижения риска, применяемая к рассматриваемому ВМ, должна быть обоснована с использованием научных подходов.
Результаты оценки рисков могут быть включены в документы регистрационного досье. Однако следует отметить, что данная оценка рисков проводится в отношении рисков безопасности и качества ВМ, и не предусматривает какой-либо оценки биологической функциональности и любого результирующего воздействия на качество и эффективность клеточного препарата.
7.1 Компоненты ВМ, их идентификация и чистота
7.1.1 Общие положения
Поставщик должен обеспечить устойчивое производство ВМ, соответствующего согласованной спецификации. Следует использовать общепринятые (фармакопейные) аналитические методики, если приемлемо. Нефармакопейные методики должны быть валидированы поставщиком.
7.1.2 Идентификация и содержание компонента(ов)
Поставщик ВМ должен описать идентификацию каждого компонента ВМ, который состоит из компонентов с определенной химической структурой. Поставщик ВМ также должен описать все компоненты с идентифицированной молекулярной структурой и их относительные концентрации для материалов, которые представляют собой смеси компонентов разной природы (химических и биологических). Поставщик ВМ должен предоставить соответствующую информацию о вариабельности партий и ее приемлемого диапазона. Поставщик ВМ и пользователь ВМ должны проводить соответствующие испытания на идентификацию. В некоторых случаях пользователю необходимо разработать другую аналитическую методику, если методика поставщика недостаточно специфична.
При наличии возможности следует обеспечить соответствие всех химических компонентов соответствующим фармакопейным требованиям.
Если идентификация всех веществ, входящих в состав ВМ, не может быть проведена и/или раскрыта, то идентификацию ВМ следует оценивать по испытанию на биологическое (функциональное) действие.
Поставщик ВМ должен описать наличие в составе ВМ любых запатентованных компонентов, по отдельности или в совокупности, а также их относительные концентрации. Должна быть предоставлена любая разрешенная для разглашения информация относительно их молекулярной структуры или назначения.
Поставщик ВМ должен подготовить CoO для материалов животного происхождения. Документированная информация должна включать страну происхождения, а также может включать заключение о состоянии здоровья животного или стада, из которых был получен данный компонент, и доказательства, подтверждающие отсутствие патогенных агентов (например, испытания на наличие вирусов, тесты на стерильность).
Для материалов человеческого происхождения требуется проведение испытаний на наличие стандартного набора вирусов. Пользователь ВМ должен учитывать соответствующие требования в стране, для которой предназначается клеточный препарат.
7.1.3 Чистота и примеси
Поставщик ВМ, состоящих из единственного компонента, который был выделен и/или очищен, должен установить в спецификации требования к чистоте этого компонента, проводить соответствующие испытания и предоставлять результаты пользователю ВМ. В случае ВМ, состоящих из нескольких компонентов, устанавливают требования и проводят испытания на чистоту активных компонентов. Для ВМ из биологических материалов со сложно определяемой структурой (например, супернатанты клеточной культуры, в которых содержится коллагеназа), следует оценивать биологическое действие, которое может негативно повлиять на эффективность таких биологических материалов в процессе производства клеточных препаратов, а также оценивать действие любого вещества, которое может вызвать неблагоприятную реакцию (например, эндотоксинов).
Если ВМ производят несколькими партиями, то должны быть проведены испытания на чистоту каждой серии ВМ. Если показатели чистоты варьируются от партии к партии в пределах допустимого диапазона, следует указать допустимый диапазон, а также информацию о вариабельности поставленных партий в пределах допустимого диапазона.
Примеси должны быть идентифицированы и включены в спецификацию, если это применимо. При необходимости, следует установить соответствующие испытания для определения содержания примесей и допустимые пределы (критерии приемлемости).
Если это возможно, поставщик ВМ должен сообщать результаты соответствующих испытаний, чтобы пользователь ВМ мог провести квалификацию клеточного препарата с целью выявления в нем остаточных количеств ВМ.
Поставщик ВМ должен продемонстрировать межсерийную однородность без указания конкретной молекулярной структуры определенных компонентов или их содержания, если информация о молекулярном составе ВМ не может быть раскрыта, например предоставляется заявление для каждого нераскрытого компонента о его межсерийной вариабельности путем указания процента его содержания в общей смеси или значение отклонения в пределах допустимого заранее установленного процентного содержания, если применимо.
Доказательства однородности состава следует предусмотреть для ВМ, содержащих неидентифицированные или нераскрытые компоненты.
Конфиденциальная информация может быть предоставлена непосредственно уполномоченному регуляторному органу (например, посредством мастер-файла на ВМ или в виде конкретного ответа на конкретный запрос в виде файла с поддерживающими регуляторными данными). Решение об объеме информации, предоставляемой поставщиком ВМ пользователю, следует обсудить и согласовать между обеими заинтересованными сторонами.
Если конфиденциальная информация о компоненте(ах) ВМ передается пользователю ВМ, то он несет ответственность за определение того, является ли материал постоянно пригодным для применения в отношении такого (таких) компонента(ов), и должен провести соответствующие подтверждающие исследования. Данные исследования могут включать, но не ограничиваться этим, испытания на контаминанты, стерильность, бактериальные эндотоксины, наличие вирусов, наличие микоплазмы, pH и осмолярность.
Если поставщик ВМ заявляет в спецификации о стерильности ВМ, метод стерилизации должен быть описан и доведен до сведения пользователя.
7.2 Хранение ВМ и его стабильность
7.2.1 Общие положения
Поставщик ВМ должен предоставить достаточный объем информации о надлежащих условиях хранения ВМ, а также описание количественных испытаний, используемых для определения стабильности продукта, соответствующую информацию о стабильности ВМ в заявленных условиях хранения.
Поставщик ВМ должен сообщать о добавлении любых стабилизаторов.
Пользователь ВМ должен проверить сохранность пригодности для использования ВМ в установленных условиях хранения.
Для определения срока годности и рекомендуемых условий хранения поставщик ВМ должен провести исследования стабильности всех форм ВМ. Показатели качества, используемые для оценки стабильности, возможно определить согласно руководствам ICH Q1 и ICH Q5C, если применимо. Методы и применимость соответствующих испытаний могут быть определены согласно руководствам ICH Q1 и ICH Q5C.
Поставщик ВМ должен предоставить пользователю ВМ следующую информацию о стабильности и условиях хранения:
a) дату производства;
b) дату окончания срока годности или срок годности при хранении.
Примечание - При необходимости может быть указан срок годности для каждого температурного диапазона замораживания (в лабораторном холодильнике или в морозильной камере глубокой заморозки) и срок хранения в холодильнике после размораживания;
c) стабильность при одной температуре или нескольких отдельных температурах, если применимо;
d) стабильность для каждой имеющейся формы ВМ;
e) рекомендуемые меры при воздействии других неблагоприятных для стабильности параметров окружающей среды, таких как воздействие интенсивного света или вибрации.
Информация о стабильности и сроках годности должна быть получена путем проведения испытаний поставщиком ВМ, и должна основываться на способности ВМ сохранять критический уровень активности или действия, а также его целостность, то есть его профиль примесей и скорость деградации, если это применимо.
Активность/действие ВМ должна оставаться в установленных пределах в течение всего срока годности ВМ при соблюдении соответствующих рекомендованных условий хранения.
Поставщик ВМ должен предоставить рекомендации по условиям хранения, например:
- температура;
- защита от света;
- влажность;
- чувствительность к pH;
- реакция на вибрации (встряхивание);
- чувствительность к воздействию окислительно-восстановительных агентов.
Если ВМ поставляется в форме или состоянии, отличном от того, в котором он будет использован (например, в виде приготовленного раствора, размороженный, аликвотированный), поставщик ВМ должен рекомендовать соответствующие условия хранения для альтернативных форм или состояний, а также предоставить данные о стабильности, при наличии. Поставщик ВМ должен предоставить процедуру приготовления раствора, а также довести до сведения пользователя ВМ информацию об изменении стабильности и других свойств, обусловленных приготовлением.
Если ВМ предполагается хранить в замороженном виде на протяжении всего транспортирования и хранения, поставщик ВМ может предоставить следующую информацию:
- влияет ли замораживание и оттаивание на стабильность и активность ВМ;
- максимальное рекомендуемое количество циклов замораживания-оттаивания;
- если применимо, указывают срок годности в каждом диапазоне температур замораживания (например, в лабораторной морозильной камере или в морозильной камере глубокого замораживания);
- если применимо, указывают срок хранения в холодильнике после размораживания.
8.1 Система менеджмента качества
Производители ВМ должны внедрить СМК. Производители ВМ должны регулярно подвергаться аудиту (см. 6.2.2). Поставщик ВМ должен внедрить процесс исправления несоответствий, который будет проверяться пользователями ВМ. Такой процесс должен включать планы выполнения эффективных корректирующих действий, то есть действий по устранению причины несоответствия и предотвращению его повторения, а также своевременное устранение выявленных критических проблем.
Примечание - ИСО 13485 и ИСО 9001 являются базовыми примерами соответствующей СМК. Необходимо оценить, достаточно ли соблюдать требования существующих стандартов на системы менеджмента, включая ИСО 9001 и ИСО 13485, в каждом конкретном случае.
Типичными элементами СМК являются:
a) общие принципы;
b) производственные участки, производственная среда;
c) производственные мощности, система поставок и снабжения;
d) системы ведения производственных записей;
e) система технического обслуживания, основанная на цикле "Планируй-делай-проверяй-действуй" (PDCA);
f) записи об образовании и внутреннем обучении персонала;
g) управление безопасностью;
h) управление поставщиками;
i) управление оборудованием;
j) управление материалами, используемыми для производства ВМ, включающая ведение записей, обеспечивающих прослеживаемость этих материалов.
Поставщики ВМ должны иметь надежную цепь поставок и возможность удовлетворять потребности пользователей ВМ.
8.2 Технологический процесс
Установленные параметры технологического процесса, влияющие на качество и однородность ВМ, должны контролироваться, документироваться и доводиться до сведения заинтересованных сторон.
Установленные параметры технологического процесса, которые могут снизить риск контаминации вирусами или эндотоксинами, следует контролировать и документировать.
Установленные параметры технологического процесса, связанные с использованием компонентов животного происхождения, должны быть доведены до сведения пользователя ВМ. В дополнение к документации о стране происхождения следует указывать меры по снижению риска контаминации вирусами.
При использовании антибиотиков следует указать их наименование и количество, присутствующих в ВМ.
Примечание - Использование бета-лактамных антибиотиков может представлять иммуногенный риск. Использование антибиотиков в производстве строго контролируют, чтобы ограничить воздействие на окружающую среду, приводящее к росту антибиотикорезистентности.
Деградация продукта в ходе производственного процесса должна быть зарегистрирована. Записи включают продукты деградации в ходе обработки и ферментативного расщепления биологических продуктов.
Количество механических включений в продукте должно быть сведено к минимуму, насколько это возможно. При наличии возможности следует предусмотреть стадии по их удалению. При наличии возможности следует включить в спецификацию испытания на размер (невидимые и видимые) и количество механических включений в ВМ.
Используемые в производственном процессе стадии химической или физической обработки для снижения контаминации или стерилизации ВМ должны быть описаны, а их эффективность продемонстрирована.
Используемая в производственном процессе стадия радиационной стерилизации должна быть описана. Диапазон поглощаемой дозы ионизирующего излучения должен быть подтвержден.
Контаминация должна контролироваться на всем протяжении технологического процесса посредством контроля производственной среды, физического разделения потоков и операций, средств технического контроля, использования выделенного "под продукт" оборудования и процедур уборки и очистки. Контаминация эндотоксинами должна быть сведена к минимуму.
Поставщик ВМ должен обеспечить целостность упаковки, чтобы гарантировать, что материалы не будут контаминированы или фальсифицированы. Необходимо валидировать пригодность специальных систем упаковки и укупорки для защиты от экстремальных температур и возможных повреждений при транспортировании.
Контейнеры и средства укупорки должны быть стерильными. При наличии рекомендуется использовать одноразовые контейнеры. Следует предоставить данные о вероятности образования экстрагируемых веществ из контейнера, выбранного пользователем ВМ.
Оценку рисков компонентов животного происхождения начинают на ранней стадии разработки клеточного препарата. Оценка рисков этих компонентов основана на риске при предполагаемом использовании.
Подходы при определении ADCF включают:
a) состав продукта: ВМ не содержит компонентов каких-либо веществ животного или человеческого происхождения (например, питательные среды для культивирования клеток, в которых в качестве основного компонента ВМ отсутствуют компоненты животного или человеческого происхождения, но используются рекомбинантные белки);
b) история производства: ВМ производят без применения материалов животного или человеческого происхождения.
Примечание - Рекомендации по риск-ориентированной системе квалификации ВМ приведены в ИСО 13022, руководствах ICH Q9, USP <1043> и [27] и [28].
Следует установить, строго соблюдать и документировать систему прослеживаемости ВМ животного или человеческого происхождения. Рекомендуется использовать доступную для поиска и быстрого извлечения информацию из базы данных и обмениваться информацией в электронном формате. Стандартный формат приведен в ASTM E3077. Все компоненты должны пройти скрининг на наличие риска передачи соответствующих заболеваний в соответствии с источником материалов. CoO, в котором указана информация об объекте, где были получены материалы, а также все соответствующие испытания и результаты должны храниться у поставщика ВМ и предоставляться по мере необходимости.
Для ВМ, содержащих компонент(ы) животного и/или человеческого происхождения, такой(ие) компонент(ы) должен (должны) быть идентифицирован(ы) и задокументирован(ы). Если ВМ содержит компоненты, которые не могут быть раскрыты, пользователю ВМ должно быть предоставлено заявление о наличии в ВМ компонента животного и/или человеческого происхождения.
ВМ, которые производятся из материалов, полученных от человека, должны производиться предприятиями, имеющими соответствующие лицензии. Такие продукты должны быть проконтролированы и документально оформлены организациями, осуществляющими донации.
8.5 Безопасность для клеток и человека
Поставщик ВМ должен обеспечить соответствие ВМ своим спецификациям, в том числе в части контаминации, включая микробную и вирусную контаминацию, контаминацию агентами небиологического происхождения и перекрестную контаминацию другими продуктами.
Поставщик ВМ должен предоставить пользователю ВМ информацию о том, как были проведены и провалидированы испытания на стерильность и процесс стерилизации ВМ, а также о количестве единиц продукции и достигнутом уровне обеспечения стерильности.
Примечание - Такая информация необходима пользователю ВМ для обеспечения безопасности и стерильности клеточного препарата, так как они не могут подвергаться стерилизации.
Пользователь должен провести дополнительные исследования ВМ, чтобы продемонстрировать его безопасность для клеток и человека. При необходимости пользователь должен оценить влияние ВМ на характеристики клеток. В зависимости от рисков, связанных с ВМ, такие исследования включают, но не ограничиваются:
a) пролиферацию клеток;
b) генотоксичность;
d) пластичность клеток;
e) миграцию клеток;
f) морфологию клеток;
g) экспрессию генов;
h) стабильность хромосом;
i) любые другие подходящие испытания.
9.1 Общие положения
Контроль характеристик ВМ может проводить как поставщик, так и пользователь. Как правило, поставщик ВМ проводит такие испытания для демонстрации соответствия ВМ общим требованиям, в то время как пользователь ВМ проводит такие испытания для подтверждения пригодности для предполагаемого использования ВМ.
Поставщик ВМ должен предоставить результаты одного или нескольких испытаний функциональных характеристик, если требуется заключение об однородности выпускаемого продукта, в частности, в отношении функциональных характеристик.
Поставщик ВМ должен проводить испытания функциональных характеристик, если небольшая вариабельность компонентов может привести к значимым изменениям взаимодействия ВМ с клетками, если это доступно (например, в случае сложных питательных сред для культивирования клеток или сывороток).
Кроме того, в случае, когда точная идентификация компонента и его концентрация неизвестны или не могут быть раскрыты, проведение пользователем ВМ испытаний функциональных характеристик может быть использовано в качестве альтернативного метода для демонстрации однородности серий клеточных препаратов.
Пользователь должен провести оценку ВМ в рамках системы квалификации, которая учитывает уровень риска, связанного с этим материалом.
Пользователь должен всесторонне оценивать ВМ на протяжении всей разработки клеточного препарата. Входе квалификации ВМ должна проводиться оценка источников происхождения, идентификации, чистоты, биологической безопасности и общей пригодности конкретного материала. Пользователь ВМ может применять риск-ориентированный подход для сокращения объемов входных испытаний при наличии соответствующего обоснования, например поставщик ВМ прошел квалификацию (включен в перечень одобренных поставщиков), сокращение объемов обосновано и проводится регулярный входной контроль качества ВМ.
Пользователь ВМ также должен провести квалификацию поставщика ВМ, то есть проверить его СМК и программы контроля материалов.
Примечание - В UPS <1043> приведены рекомендации по разработке системы квалификации ВМ, основанной на оценке рисков.
При возможности в качестве ВМ следует использовать высококачественные материалы с подтвержденными показателями качества.
9.3 Проведение квалификации ВМ
Пользователь должен выбрать ВМ и поставщиков ВМ исходя из используемого процесса производства клеточных препаратов.
Пользователь ВМ должен применять поэтапный подход к выбору ВМ, который обеспечивает баланс между повышающимися требованиями по снижению рисков и стоимостью сдерживания биологических опасностей на различных этапах разработки клеточного препарата.
Пользователь ВМ должен разработать спецификацию для каждого ВМ, чтобы обеспечить однородность серий и эффективность процесса производства клеточного препарата. Пользователь ВМ должен валидировать методики испытаний, включенные в эти спецификации. Рекомендации по проведению надлежащей валидации приведены в ИСО 5725-1 - ИСО 5725-5, ICH Q2(R1) и ИСО/МЭК 17025.
Если пользователь ВМ намеревается использовать ВМ не в соответствии со спецификациями поставщика ВМ, он должен установить для ВМ предполагаемые условия хранения, спецификации и срок годности.
При внесении изменений в процесс производства ВМ или клеточного препарата может потребоваться оценка рисков новых процессов и информирование соответствующих сторон.
Программа КК пользователя ВМ, такая как в GMP, должна подтверждать текущие показатели качества каждого ВМ в его предполагаемом применении посредством разработанного и внедренного плана квалификации ВМ. Типичная стратегия КК также должна охватывать:
a) управление поступающими материалами путем приемки, отдельного хранения, проведения испытаний и разрешения для использования в производстве (перед использованием);
b) соглашения о качестве и поставках, заключенные между поставщиком ВМ и пользователем ВМ;
c) постоянную оценку однородности серий и анализ воздействия на готовый продукт;
d) аудит и квалификацию продавца (поставщика);
f) политику и практику действий в отношении материалов, не соответствующих спецификации;
g) программу исследования стабильности (результаты исследований может предоставить поставщик ВМ);
h) порядок отбора проб (например, удвоение плана отбора проб при необходимости проведения расследования);
i) хранение архивных образцов;
j) CoO (если применимо);
k) условия транспортирования и хранения (условия должны быть подтверждены).
Пользователь ВМ должен определить, задокументировать и внедрить стратегии КК при производстве клеточных препаратов в системе менеджмента, соответствующей требованиям настоящего стандарта <*>.
--------------------------------
<*> Фармацевтическая система качества должна также соответствовать обязательным требованиям к производству и изготовлению клеточных препаратов.
9.4 Испытания функциональных показателей
Поставщик ВМ должен разработать и валидировать методики оценки функциональных показателей для демонстрации действия ВМ на клетки и/или однородности серий ВМ. Испытания могут включать в себя оценку стандартных функций клеток (например, выживаемости клеток, пролиферации), или исследования более специфических показателей идентификации и функций клеток (например, экспрессия клеточно-специфических генов или количественное определение функциональной активности клеток).
Пользователь ВМ должен выбирать испытания функциональных показателей исходя из предназначения ВМ. Поставщик ВМ должен предоставлять пользователю ВМ достаточные данные о методах испытаний, чтобы пользователь мог оценить применимость полученных результатов к использованию ВМ.
9.5 Результаты испытаний функциональных показателей
Результаты испытаний функциональных показателей для использования по назначению должны документироваться пользователем ВМ. Результаты общих испытаний функциональных показателей, выполненных поставщиком ВМ, также могут быть предоставлены пользователю ВМ. Результаты, если возможно, должны быть представлены в таком виде, чтобы способствовать выбору надлежащего ВМ и/или оценке соответствия их целевому назначению.
По запросу может быть предоставлен результат, демонстрирующий однородность серий ВМ. Результаты испытаний функциональных показателей могут включать:
a) количественные результаты;
b) распределение партий в пределах приемлемого диапазона;
c) неопределенность количественного результата при использовании биологических методов.
Поставщик и пользователь ВМ должны совместно определять достаточный объем данных. Баланс между объемом информации и специфичностью испытания функциональных показателей должен обеспечивать достаточный уровень доказательств однородности серий в отношении пригодности для применения.
10.1 Общие требования
Так как ВМ предназначены для производства клеточных препаратов в соответствии с настоящим стандартом, поставщики ВМ должны информировать об этом предназначении пользователя ВМ путем указания соответствующей информации на этикетке и/или в каталоге, например, "предназначен для использования в производстве препаратов клеточной терапии или генной терапии".
Примечание - Пригодность ВМ для использования, как правило, определяет пользователь ВМ.
Поставщик ВМ должен предоставить достаточную информацию о результатах оценки качества, чтобы обеспечить общее понимание и возможность сравнения данных. Информация должна включать, но не ограничиваться:
a) первичные данные;
b) обработанные данные;
c) шкалу(ы) для количественных или относительных данных;
d) сравнение с известным(и)/эталонным(и) значением(ями);
e) метаданные (касающиеся образцов, процедур, условий и т.п.);
f) результаты стратегий контроля (для обеспечения доверия к проведенному испытанию);
g) функциональные характеристики продукции;
h) стабильность;
i) КК;
j) аналитические методики.
Поставщик ВМ несет обязательства по предоставлению CoA и CoO. Поставщик ВМ должен иметь в виду, что предоставленная им информация оценивается пользователем ВМ и что в некоторых случаях поставщику ВМ может быть необходимо оказать помощь при проведении аудита пользователем ВМ по предоставлению подробной информации. Информация, предоставленная поставщиком ВМ, также может быть использована для установления контакта (подписки на получение информации), чтобы пользователь ВМ мог получать любую доступную новую информацию от поставщика ВМ. Рекомендуется, чтобы поставщик ВМ и пользователь ВМ заключили соглашение об обмене информацией в ходе предварительного обсуждения и взаимного согласования, что должно быть отражено в соглашении по качеству.
Этапы коммерциализации начинаются только после соглашения об аспектах GMP для ВМ. Следование GMP укрепляет партнерские отношения между поставщиком ВМ и пользователем ВМ.
SDS должен предоставляться для всех веществ и смесей, например, соответствующих критериям физической опасности, опасности для здоровья или опасности для окружающей среды, описанным в [29], а также для всех смесей, содержащих компоненты, соответствующие критериям канцерогенности, репродуктивной токсичности или токсичности в отношении органов-мишеней в концентрациях, превышающих соответствующие задокументированные предельные значения. Информация должна включать положения о любых запретах или ограничениях, которые могут применяться в зависимости от страны или региона, в который поставляется ВМ.
Предоставляемая о ВМ информация должна включать:
- заголовок (например, "Отчет по качеству" или "Сертификат на материал");
- идентификатор ВМ, описание клеток, из которых получен ВМ (например, тип клетки, номер серии или идентификационный номер, дата производства);
- наименование и адрес организации-производителя, а также место, где проводились испытания, указанные в отчете, если они отличаются от адреса организации-производителя;
- дату публикации отчета в стандартном формате в соответствии с ИСО 8601-1;
- уникальную идентификацию отчета (например, серийный номер), который указывается на каждой странице для демонстрации ее принадлежности к отчету, а также в конце отчета;
- биологическую идентификацию материала или особые свойства (если существует биологический источник ВМ);
- описание аналитических методик, включая их чувствительность, специфичность и критерии приемлемости;
- результаты испытаний с указанием, если приемлемо, единиц измерения.
Примечание - Единицами измерения могут быть единицы СИ или другие единицы, определенные для целей информирования;
- данные о произошедших отклонениях.
Кроме того представляемая информация может включать:
- соответствующую информацию о качестве ВМ и связанных с ним данных;
- условия хранения;
- дату окончания срока годности.
Поставщик должен предоставить CoA ВМ. Если в процессе разработки потребуется заменить ВМ, то следует использовать эквивалентный ВМ, что должно быть научно обосновано и соответствовать регуляторным требованиям.
Примечание - CoA позволяет сравнивать ВМ для принятия обоснованного решения при замене. Это подчеркивает необходимость установить робастные показатели качества ВМ. Рекомендации по установлению показателей качества ВМ приведены в приложении D.
Ко всем ВМ должен прилагаться CoA, содержащий, в зависимости от обстоятельств, количество, номер серии, результаты испытаний конкретной партии и срок годности, при этом испытания должны включать (допускается более одного испытания для каждого показателя качества) следующее:
a) идентификация:
- методы идентификации должны обладать как можно большей специфичностью (например, идентификация последовательности N-концевого белка или соответствующее испытание идентификации материала, защищенного интеллектуальной собственностью, такого как питательной среды для культивирования клеток);
b) количественное определение (содержание):
- методы количественного определения должны быть как можно более специфичными (например, специфическая абсорбция белков);
- удельная плотность некоторых жидкостей, позволяющая измерять их количество путем взвешивания, в дополнение к объему;
c) чистота и примеси:
- количественные испытания на наличие соответствующего материала и потенциальных контаминантов (например, SDS-PAGE, RP-HPLC);
d) безопасность:
- для проведения испытаний на безопасность следует использовать фармакопейные методики и/или валидированные методики с использованием образцов в количестве, достаточном для подтверждения соответствия применимым требованиям. Испытания могут включать определение стерильности, эндотоксинов, микоплазмы и вирусов в зависимости от материалов и потенциальной безопасности;
e) функциональные показатели/биологическая активность:
- если ВМ выполняет биологическую функцию, биологическую активность следует измерять с помощью количественного измерения биологической функции. Активность может быть выражена в виде специфической активности. Аналитическая система должна калиброваться с помощью признанного международного стандартного образца (стандартных материалов), если таковой имеется, например, Всемирной организации здравоохранения, и иметь обоснованный определенный рабочий диапазон. В тех случаях, когда это невозможно, измерения биологической активности должны проводиться с помощью соответствующих и надежных методик, а ограничения методик должны быть достаточно изучены;
f) биохимические показатели;
g) осмоляльность;
h) показатель pH.
10.4 Дополнительные сертификаты
Поставщик ВМ должен управлять цепочкой поставок всех материалов и предоставлять сертификаты о происхождении, особенно для материалов животного или человеческого происхождения, включая сыворотку, трипсин, молоко и другие биологические жидкости или их производные. В CoO должны быть включены подробные сведения, такие как информация о ADCF, описанная в 8.4.
10.4.2 Сертификат соответствия
При наличии возможности поставщик ВМ может предоставить документы, подтверждающие соответствие требованиям систем качества или стандартам.
10.4.3 Сертификат радиационной обработки
Если это возможно, поставщик ВМ может предоставить CoI, когда это применимо (на материалы, подвергшиеся радиационной стерилизации, такие как эмбриональная бычья сыворотка; или о других мерах по снижению вирусной активности).
11.1 Влияние изменений на ВМ
В некоторых случаях поставщику ВМ необходимо внести изменения в ВМ, чтобы обеспечить требуемое качество препарата клеточной терапии. Необходимость обеспечения постоянства качества ВМ требует особого внимания, если поставщик ВМ:
a) вносит изменения в процесс производства (включая состав биологического ВМ);
b) прекращает производство;
c) заменяет производственную площадку.
Подходы по внесению изменений в ВМ приведены в руководстве ICH Q12.
Определенные изменения технологического процесса или состава ВМ биологического происхождения, используемого на любом этапе жизненного цикла, могут повлиять на качество препарата клеточной терапии. Это может нарушить валидность исследований, проведенных до внесения изменений.
Поставщик ВМ должен определить тип каждого изменения и оценить, требуется ли уведомление о нем пользователя ВМ, например в законодательстве ряда стран установлены типы изменений ВМ и соответствующие процедуры информирования о них регуляторных органов (критические, значительные, незначительные и т.д.), и не обо всех изменениях требуется уведомлять регуляторный орган. Необходимо сообщать об изменениях биологического контроля за ВМ, которые влияют на качество, безопасность или эффективность производимого ВМ. Оценка изменений, о которых необходимо сообщать, как правило, является более гибкой при разработке, чем при промышленном производстве.
При внесении изменений в ВМ (например, изменений в технологический процесс ВМ, состав, упаковку, упаковочную систему и/или укупорочное средство) поставщик ВМ должен уведомить пользователя ВМ о таких изменениях как можно скорее или заблаговременно, пока эти изменения не начали действовать. Поставщик ВМ и пользователь ВМ должны заключить соглашение по качеству, которое будет содержать это требование.
Одновременно пользователь ВМ должен определить и провести квалификацию альтернативного поставщика биологического ВМ, поскольку предпочтительный поставщик ВМ может прекратить его производство. Таким образом, соглашение по качеству, заключенное пользователем ВМ, должно содержать четкий раздел, касающийся непрерывности поставок.
Пользователь ВМ должен иметь соответствующую документацию, позволяющую проводить исследования по сопоставимости с эквивалентным или модифицированным биологическим ВМ. Пользователь ВМ должен включить в первоначальное соглашение с поставщиком ВМ разделы, предусматривающие, что в случае внесения изменений в производственный процесс этого продукта поставщик ВМ должен своевременно уведомить об этом пользователя ВМ, и указать требуемый срок уведомления.
(справочное)
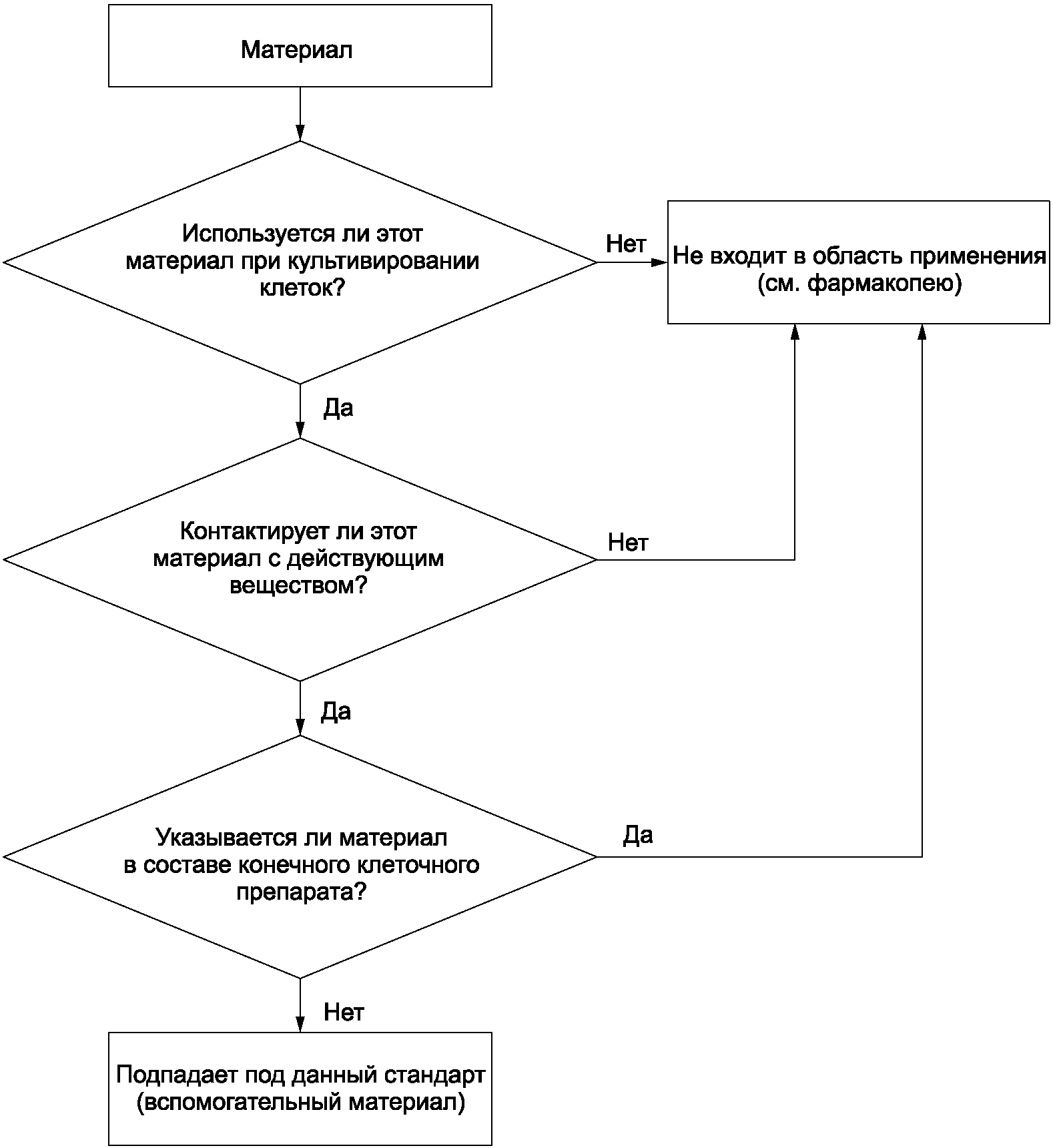
Рисунок A.1 - Блок-схема принятия решения по включению
материала в область применения настоящего стандарта
В настоящем стандарте к ВМ относят как минимум следующее:
a) реактивы;
b) антикоагулянты;
c) цитокины;
d) факторы роста;
e) ферменты;
f) антитела;
g) сыворотку (человеческую или крупного рогатого скота);
h) буферные растворы;
i) питательные среды;
j) чашки (покрытые биологическим материалом);
k) частицы (покрытые биологическим материалом);
l) криопротекторы (средства для криоконсервации);
m) активирующие агенты/реактивы;
n) клетки немлекопитающих (например, клетки насекомых, бактериальные клетки);
o) плазмиды <*>;
p) вирусные векторы <*>.
--------------------------------
<*> При применении настоящего стандарта следует учитывать, что в соответствии с Решением Совета ЕЭК N 89 от 3 ноября 2016 г. вирусные векторы и плазмиды, применяемые для трансфекции или других целей, относятся к исходным материалам независимо от того, входят ли они в состав готового продукта. Производство и контроль качества таких веществ осуществляется в соответствии с принципами надлежащей производственной практики (GMP), утвержденных Решением Совета ЕЭК N 77 от 3 ноября 2016 г.
В настоящем стандарте к ВМ не относят:
- клетки, которые являются исходными материалами, промежуточными продуктами или готовой формой препарата клеточной терапии;
- питающие клетки;
- добавки, используемые после завершения культивирования клеток;
- каркасы;
- небиологические расходные материалы [например, стеклошарики (бусы), чашки, колбы для культивирования тканей, пакеты, трубки, пипетки, иглы];
- другие пластмассовые принадлежности и посуду, контактирующие с клеткой или тканью;
- оборудование;
- приборы.
(справочное)
ПОСТАВЩИКА ВМ И ПОЛЬЗОВАТЕЛЯ ВМ
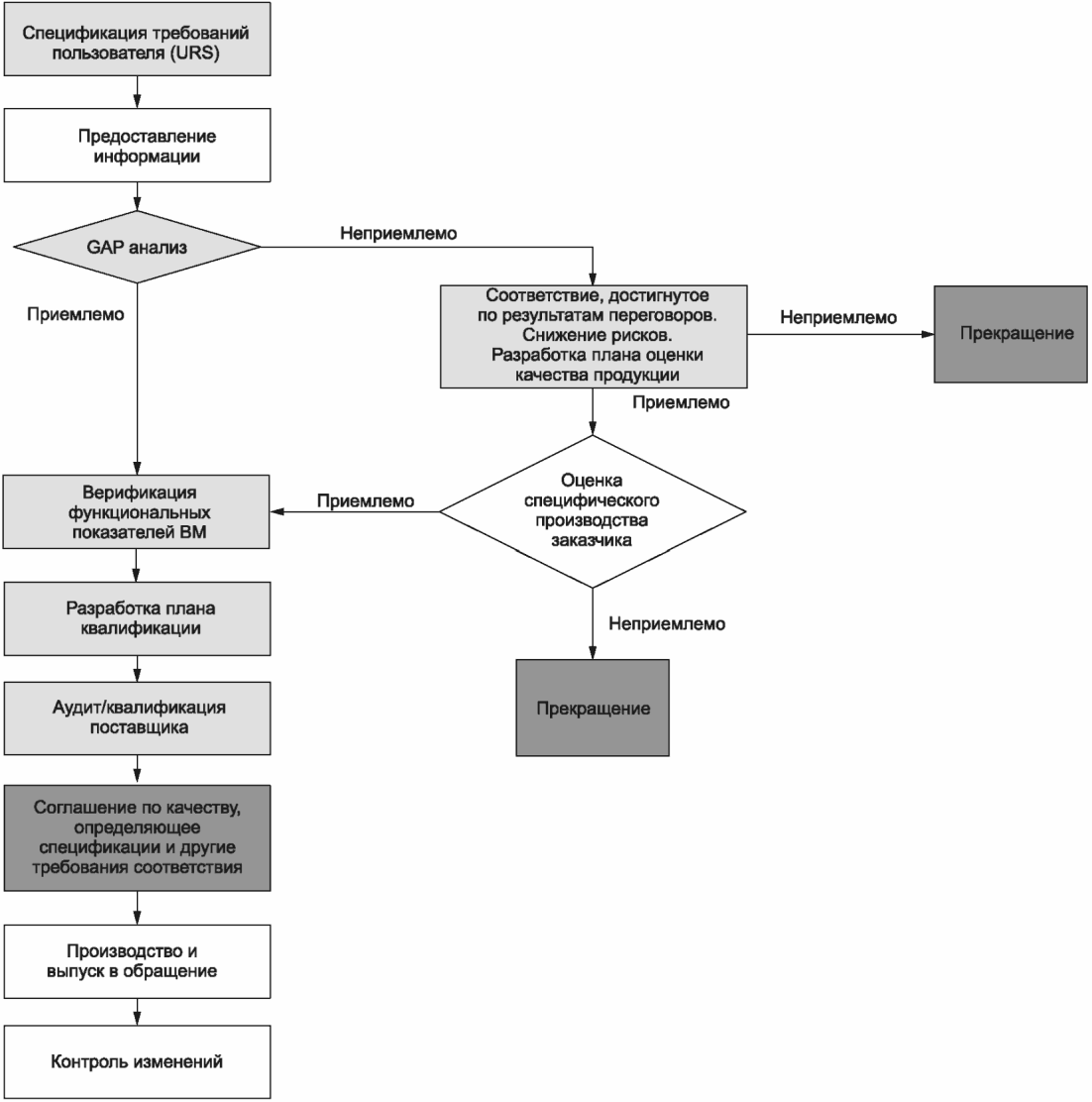
Рисунок B.1 - Пример блок-схемы взаимодействия
поставщика ВМ и пользователя ВМ
(справочное)
Следующая информация может быть предоставлена в открытых для общего пользования документах или документах ограниченного распространения:
Примечание - Предоставление конфиденциальной информации может быть предметом соглашения о взаимном раскрытии конфиденциальной информации.
a) информация о ВМ, которую указывают и предоставляют в одном или нескольких технических документах, соглашении о поставках или мастер-файл для ВМ:
1) краткое описание продукта:
i) наименование продукта (наименование ВМ);
ii) каталожный номер;
iii) условия хранения и максимальный срок годности;
iv) производитель;
v) наименование источника импорта и наименование брокера (если применимо);
2) сведения о качестве:
i) спецификация и аналитическая методика;
3) компоненты:
i) материалы компонентов;
ii) концентрация (содержание) компонентов;
4) сведения о компонентах, используемых в производстве ВМ:
i) присутствие или отсутствие компонентов человеческого или животного происхождения (см. 5.2);
ii) присутствие опасных материалов, если применимо;
iii) компонент, содержащий тяжелые металлы (см. например, BSI PAS 157:2015, классы 1, 2a, 2b, 3);
iv) страна происхождения;
5) информация, необходимая при использовании материалов биологического происхождения в производстве ВМ:
i) безопасность в части патогенных агентов, включая вирусы и TSE/BSE.
Пример 1 - Испытания при аттестации банка клеток, испытания, подтверждающие удаление или инактивацию вируса, валидация процесса инактивации вирусов, если применимо, в сыворотках или плазме крови человека или продуктов животного происхождения;
ii) результаты испытаний на микробиологическую чистоту;
iii) страна происхождения;
6) соглашение по качеству;
i) история разработки (вероятно, она будет включена только в досье);
b) информация о серии ВМ и поставляемая вместе с серией:
1) CoA и CoO:
i) краткое описание продукта;
ii) наименование продукта (наименование ВМ);
iii) каталожный номер;
iv) номер серии/партии;
v) условия хранения;
vi) производитель;
vii) срок годности;
viii) наименование источника импорта;
ix) наименование брокера или дистрибьютора, в зависимости от обстоятельств;
x) спецификация и результаты испытаний;
xi) информация о компонентах, используемых для производства ВМ;
xii) присутствие или отсутствие компонентов человеческого или животного происхождения (см. 5.2);
xiii) присутствие опасных материалов, если применимо;
xiv) страна происхождения;
xv) безопасность в части патогенных агентов, включая вирусы и TSE/BSE.
Пример 2 - Испытания при аттестации банка клеток, испытания, подтверждающие удаление или инактивацию вирусов;
xvi) результаты испытаний на микробиологическую чистоту;
xvii) маркировка поставляемой серии;
xviii) предполагаемое использование продукта.
(справочное)
Пользователь ВМ и/или поставщик ВМ могут провести испытания и/или характеризацию, чтобы убедиться, что качество ВМ соответствует их критериям. Однако после того, как пользователь ВМ одобрил ВМ, ответственность за дальнейший контроль качества и характеризацию этого материала и всех связанных с этим документов лежит на пользователе ВМ. Как правило, ВМ может быть подвергнут различным испытаниям для получения дополнительной информации по ряду показателей, но объем этих испытаний зависит от объема контроля, проводимого поставщиком ВМ, и валидности предоставляемых им данных. Краткий перечень показателей включает:
a) внешний вид: краткое описание того, как выглядит ВМ;
b) идентификацию: испытания на подтверждение присутствия представляющего интерес вещества (аналита);
c) чистоту: испытание на чистоту вещества;
d) примеси, которые подразделяются:
1) на примеси ВМ: типичные продукты деградации ВМ, но в случае использования биологических материалов примеси ВМ могут включать другие нежелательные вещества, которые входили в состав исходного материала.
Примечание - Такой подход применяют при наличии доступной технологии для эффективной оценки биологических материалов;
2) примеси, образующиеся в ходе технологического процесса: остаточные количества после технологического процесса;
3) контаминанты: любые патогенные агенты, попадающие в технологический процесс с исходным материалом, как правило, бактериальные (стерильность, эндотоксины) и вирусные агенты, также к ним могут быть отнесены связанные с процессом примеси (растворители, вымываемые и высвобождаемые вещества и т.п.);
e) содержание (количественное): количество/концентрация соответствующего вещества в первичной упаковке;
f) биологическая активность: если материал ВМ является биологически активным, например активность определяется по активности ферментов и единицам цитокиновой активности (необходимо использовать общепринятые стандартные образцы и материалы или собственный стандартизованный материал, если стандартный образец или материал недоступен);
g) другие соответствующие характеристики, например pH, осмоляльность, электропроводность, объем, масса, плотность, содержание воды, видимые и невидимые механические включения.
(справочное)
ИСПОЛЬЗУЕМЫХ ПРИ ПРОИЗВОДСТВЕ КЛЕТОЧНОГО ПРЕПАРАТА
Поставщики могут использовать ряд различных деклараций о качестве биологических препаратов, которые они поставляют.
В таблице E.1 представлен перечень деклараций о качестве от поставщиков ВМ, в которых подтверждается, что ВМ одобрен регуляторным органом и/или соответствует признанному уровню качества.
Таблица E.1
используемых поставщиками материалов ВМ, применяемых
в производстве клеточных препаратов
Декларация качества | Техническое и регуляторное описание |
Зарегистрированный терапевтический препарат или лекарственное средство | По определению, зарегистрированный терапевтический препарат или лекарственный препарат обладает требуемым качеством, безопасностью и эффективностью для заявленного применения и, следовательно, может рассматриваться как наивысший стандарт качества |
Фармакопейное качество | Если ВМ, используемый при производстве клеточного препарата, описан в соответствующей фармакопее, пользователь может сослаться на соответствующую фармакопейную статью для подтверждения соответствия уровню качества, который считается достаточным для использования при производстве этого клеточного препарата. В такой статье могут быть приведены более общие показатели для разных групп ВМ, но они не являются показателями качества, относящимися к какому-либо конкретному ВМ |
Фармацевтическая категория | Питательные среды для культивирования клеток могут соответствовать критериям отнесения к медицинским изделиям класса II <*> (средний риск) и, следовательно, могут быть сертифицированы как продукция для фармацевтического применения в соответствующей процедуре допуска медицинского изделия на рынок. В соответствии с этим положением пользователи могут получить разрешение на использование медицинского изделия, продемонстрировав его существенную эквивалентность изделию класса II <*> которое уже прошло оценку (сертификацию, регистрацию и др.). Уведомление о фармацевтической категории свидетельствует о том, что продукт был изготовлен в условиях действующей СМК |
Сертификат соответствия требованиям Европейской фармакопеи EDQM (CEP) | Для того чтобы поставщик биологического ВМ, который надлежащим образом контролируется уполномоченными органами (в данном контексте), получил сертификат CEP, поставщик предоставляет подробную информацию о своих процессах и процедурах, а EDQM подтверждает, что они снижают риск передачи TSE. Сертификат CEP признается во всех странах ЕС и ряде стран, не входящих в ЕС, таких как Канада, Новая Зеландия и Австралия |
--------------------------------
<*> В соответствии с Приказом Минздрава N 4Н от 6 июня 2012 г. регистрации в качестве медицинского изделия подлежат питательные среды, используемые для диагностики in vitro, и могут иметь степень риска 1 или 2a.
Вопросы, которые следует учитывать при декларировании качества, включают как минимум следующее:
a) соответствие какой-либо категории и/или стандарту, не обязательно означает, что ВМ пригоден для конкретного технологического процесса, даже если пригодность по его качеству очевидна. Пользователь ВМ несет ответственность за обеспечение того, чтобы продукт обладал необходимыми свойствами (например, функциональными показателями и профилем безопасности) посредством документированного процесса квалификации;
b) единое мнение о значении часто используемых условиях декларирования качества отсутствует. Пользователь ВМ несет ответственность за полную проверку квалификации ВМ независимо от заявленной категории ВМ. Также в обязанности пользователя ВМ входит получение информации о порядке присвоения соответствующих категорий ВМ, проведения оценки и управления рисками ВМ. Рекомендации по этим вопросам приведены в ICH Q9, USP <1240> и в [30];
c) следует тщательно оценивать или квалифицировать ВМ с декларированным качеством с точки зрения их качества, безопасности и пригодности. В частности, следует учитывать наличие или использование компонентов животного или человеческого происхождения в составе ВМ или в качестве производственного материала, считающегося ADCF, AOF или не содержащего ксеногенных агентов (см. 8.4).
(справочное)
НАЦИОНАЛЬНЫМ СТАНДАРТАМ
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ISO 8601-1 | - | |
[1] | ISO 9000:2015 | Quality management systems - Fundamentals and vocabulary (Системы менеджмента качества. Основные положения и словарь) |
[2] | ISO 9001 | Quality management systems - Requirements (Системы менеджмента качества. Требования) |
[3] | ISO 5725-1 | Accuracy (trueness and precision) of measurement methods and results. Part 1: General principles and definitions [Точность (правильность и прецизионность) методов и результатов измерений. Часть 1. Общие принципы и определения] |
[4] | ISO 5725-2 | Accuracy (trueness and precision) of measurement methods and results. Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method [Точность (правильность и прецизионность) методов и результатов измерений. Часть 2. Основной метод определения повторяемости (сходимости) и воспроизводимости стандартного метода измерений] |
[5] | ISO 5725-3 | Accuracy (trueness and precision) of measurement methods and results. Part 3: Intermediate precision and alternative designs for collaborative studies [Точность (правильность и прецизионность) методов и результатов измерений. Часть 3. Промежуточные показатели прецизионности стандартного метода измерений] |
[6] | ISO 5725-4 | Accuracy (trueness and precision) of measurement methods and results. Part 4: Basic methods for the determination of the trueness of a standard measurement method [Точность (правильность и прецизионность) методов и результатов измерений. Часть 4. Основные методы определения правильности стандартного метода измерений] |
[7] | ISO 5725-5 | Accuracy (trueness and precision) of measurement methods and results. Part 5: Alternative methods for the determination of the precision of a standard measurement method [Точность (правильность и прецизионность) методов и результатов измерений. Часть 5. Альтернативные методы определения прецизионности стандартного метода измерений] |
[8] | ISO 13022 | Medical products containing viable human cells - Application of risk management and requirements for processing practices (Медицинская продукция, содержащая жизнеспособные человеческие клетки. Применение менеджмента риска и требования к методикам обработки) |
[9] | ISO 13485 | Medical devices - Quality management systems - Requirements for regulatory purposes (Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования) |
[10] | ISO 20387:2018 | Biotechnology - Biobanking - General requirements for biobanking (Биотехнология. Биобанкинг. Общие требования к биобанкингу) |
[11] | ISO 35001:2019 | Biorisk management for laboratories and other related organisations (Менеджмент биорисков для лабораторий и других родственных организаций) |
[12] | ISO Guide 30:2015 | Reference materials - Selected terms and definitions (Стандартные образцы. Избранные термины и определения) |
[13] | ISO Guide 73:2009 | Risk management - Vocabulary (Менеджмент риска. Словарь) |
[14] | ISO/IEC 17025 | General requirements for the competence of testing and calibration laboratories (Общие требования к компетентности испытательных и калибровочных лабораторий) |
[15] | ISO/IEC Guide 99 | International vocabulary of metrology - Basic and general concepts and associated terms (VIM) [Международный словарь по метрологии. Основные и общие понятия и связанные с ними термины (VIM)] |
[16] | ASTM E3077 | Standard Guide for Raw Material eData Transfer from Material Suppliers to Pharmaceutical & Biopharmaceutical Manufacturers (Стандартное руководство по передаче данных eData о сырье от поставщиков материалов производителям фармацевтических и биофармацевтических препаратов) |
[17] | BSI PAS 157:2015 | Evaluation of materials of biological origin used in the production of cell-based medicinal products guide (Руководство по оценке материалов биологического происхождения, используемых при производстве лекарственных средств на клеточной основе) |
[18] | ICH Q1 | Stability testing of new drug substances and drug products |
[19] | ICH Q2(R1) | Validation of analytical procedures: text and methodology |
[20] | ICH Q5A(R1) | Viral safety evaluation of biotechnology products derived from cell lines of human or animal origin |
[21] | ICH Q5C | Quality of Biotechnological Products Stability Testing of Biotechnological/Biological Products |
[22] | ICH Q9 | Quality Risk Management |
[23] | ICH Q12 | Technical and regulatory considerations for pharmaceutical product lifecycle management |
[24] | USP <1043> Ancillary Materials for Cell, Gene, and Tissue-Engineered Products | |
[25] | USP <1240>, Virus Testing of Human Plasma for Further Manufacture | |
OIE. World organization for animal health, http://www.oie.int/ | ||
APIC Dec 2009 Supplier Qualification and Management Guideline | ||
Rathore AS, Kumar D, Kateja N. Role of raw materials in biopharmaceutical manufacturing: risk analysis and fingerprinting. Curr Opin Biotechnol. 2018 Oct; 53:99-105. doi: 10.1016/j.copbio.2017.12.022. Epub 2018 Jan 4. PMID: 29306677 | ||
UNECE, Globally Harmonized System of Classification and Labelling of Chemicals (GHS), GHS. (Rev. 7) (2017) | ||
MHLW PFSB/ELD No. 1002-1, PFSB/DC No. 1002-5, October 2, 2014, Implementation of the Standards for Biological Ingredients | ||
УДК 615.07:006.354 | ОКС 07.080 |
Ключевые слова: биотехнология, вспомогательные материалы, поставщик вспомогательного материала, пользователь вспомогательного материала, клеточный препарат | |