СПРАВКА
Источник публикации
М.: ФГБУ "Институт стандартизации", 2025
Примечание к документу
Документ вводится в действие с 01.06.2026.
Взамен ГОСТ Р ИСО 13408-2-2007.
Название документа
"ГОСТ Р ИСО 13408-2-2025. Национальный стандарт Российской Федерации. Асептическое производство медицинской продукции. Часть 2. Стерилизующая фильтрация"
(утв. и введен в действие Приказом Росстандарта от 09.06.2025 N 540-ст)
"ГОСТ Р ИСО 13408-2-2025. Национальный стандарт Российской Федерации. Асептическое производство медицинской продукции. Часть 2. Стерилизующая фильтрация"
(утв. и введен в действие Приказом Росстандарта от 09.06.2025 N 540-ст)
Содержание
Приказом Федерального
агентства по техническому
регулированию и метрологии
от 9 июня 2025 г. N 540-ст
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
АСЕПТИЧЕСКОЕ ПРОИЗВОДСТВО МЕДИЦИНСКОЙ ПРОДУКЦИИ
ЧАСТЬ 2
СТЕРИЛИЗУЮЩАЯ ФИЛЬТРАЦИЯ
Aseptic processing of health care products.
Part 2. Sterilizing filtration
(ISO 13408-2:2018, IDT)
ГОСТ Р ИСО 13408-2-2025
ОКС 11.080.01
Дата введения
1 июня 2026 года
1 ПОДГОТОВЛЕН Федеральным государственным бюджетным учреждением "Российский институт стандартизации" (ФГБУ "Институт стандартизации") на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 458 "Разработка, производство и контроль качества лекарственных средств"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 9 июня 2025 г. N 540-ст
4 Настоящий стандарт идентичен международному стандарту ИСО 13408-2:2018 "Асептическое производство медицинской продукции. Часть 2. Стерилизующая фильтрация" (ISO 13408-2:2018 "Aseptic processing of health care products - Part 2: Sterilizing filtration", IDT).
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные и межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА.
Дополнительные сноски в тексте стандарта, выделенные курсивом, приведены для пояснения текста оригинала
5 ВЗАМЕН ГОСТ Р ИСО 13408-2-2007
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ "О стандартизации в Российской Федерации". Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.rst.gov.ru)
ИСО 13408-1 устанавливает общие требования к асептическому производству медицинской продукции. Поскольку дополнительные требования для таких процессов, как: стерилизующая фильтрация, лиофилизация, очистка и стерилизация на месте, изолирующие системы и альтернативные процессы при производстве медицинских изделий и комбинированной медицинской продукции, - являются достаточно объемными для приведения их в качестве приложений в ИСО 13408-1, было принято решение о разработке дополнительных частей ИСО 13408. С дополнительными требованиями, установленными для каждого из перечисленных процессов, можно подробнее ознакомиться в частях 2 - 7 ИСО 13408.
Стерилизующая фильтрация является важнейшим этапом асептического технологического процесса. Валидация процесса стерилизующей фильтрации может быть комплексной, поскольку, как правило, ее проводят для конкретного процесса, используемого для конкретного продукта. Настоящий стандарт устанавливает требования, при соблюдении которых процесс стерилизующей фильтрации обеспечивает эффективное удаление микроорганизмов из фильтруемой среды (жидкости или газа) и получение качественного фильтрата. Соблюдение установленных требований способствует обеспечению надежности и воспроизводимости стерилизующей фильтрации и таким образом позволяет с достаточной уверенностью заявлять о том, что используемый(ые) стерилизующий(ие) фильтр(ы) при заданных условиях работы обеспечивает(ют) получение стерильного фильтрата. Воспроизводимость и надежность процесса стерилизующей фильтрации являются важными характеристиками, поскольку задерживание микроорганизмов и физическая целостность стерилизующего фильтра не могут контролироваться непрерывно в течение всего процесса фильтрации, в отличие от стерилизации с использованием биоцидов, где характеристики процесса могут контролироваться на протяжении всего процесса.
Если в процессе валидации установлена воспроизводимая взаимосвязь между способностью стерилизующего фильтра задерживать микроорганизмы, специфичные для фильтруемой среды, и физической целостностью данного фильтра, то используют подходящие неразрушающие испытания на целостность фильтра до и после его использования, чтобы определить, успешно ли прошел процесс фильтрации произведенной серии продукции. При терминальной стерилизации кинетика инактивации подчиняется математической зависимости, что позволяет рассчитать гарантированный уровень стерильности (SAL <*>). При удалении микроорганизмов из фильтруемой среды путем фильтрации данный математический закон не соблюдается, поэтому для продукта, стерилизованного путем фильтрации, термин "гарантированный уровень стерильности" не применим.
--------------------------------
С момента публикации первого издания настоящего стандарта произошло значительное развитие в области разработки и использования биофармацевтических препаратов, медицинских изделий биологического действия и биомедицинских клеточных продуктов. В данном издании настоящего стандарта подчеркивается важность глубокого понимания природы микроорганизмов, составляющих бионагрузку на стерилизуемую фильтруемую среду, включая ее взаимосвязь с типовыми микроорганизмами, используемыми для определения эффективности стерилизующего фильтра. Например, контаминация микоплазмой может стать серьезной проблемой при производстве биофармацевтических, биотехнологических и биомедицинских клеточных продуктов. Глубокое понимание аспектов, связанных с реальной биологической нагрузкой, позволит применять соответствующие меры предосторожности при разработке, валидации и контроле процесса стерилизующей фильтрации для обеспечения безопасности и качества фильтрата.
Нумерация положений в настоящем стандарте осуществлена для удобства применения пользователем стандарта и не предполагает, чтобы они выполнялись в том порядке, в котором представлены. Положения настоящего стандарта не обязательно применяют последовательно, поскольку программы разработки и валидации могут быть циклическими. Для выполнения некоторых положений могут быть привлечены отдельные лица и/или организации. Настоящий стандарт не устанавливает конкретные лица или организации, которые должны выполнять соответствующие положения.
Рекомендации по применению настоящего стандарта приведены в приложении А.
Настоящий стандарт устанавливает требования к стерилизующей фильтрации как этапу асептического производства медицинской продукции, осуществляемому в соответствии с ИСО 13408-1. Стандарт также устанавливает рекомендации по соблюдению требований к монтажу, валидации и порядку проведения процесса стерилизующей фильтрации.
Настоящий стандарт не применим к процессам удаления вирусов.
Стерилизующая фильтрация не применима к фильтруемым средам, содержащим частицы размером, превышающим размер пор фильтра (например, бактериальные цельноклеточные вакцины).
Настоящий стандарт не распространяется на высокоэффективные фильтры очистки воздуха (HEPA-фильтры).
Настоящий стандарт не устанавливает требований к разработке, валидации и контролю процесса удаления возбудителей губчатых энцефалопатий, таких как почесуха, губчатая энцефалопатия крупного рогатого скота и болезнь Крейтцфельдта-Якоба. В отдельных странах разработаны особые рекомендации по обработке материалов, потенциально зараженных перечисленными возбудителями.
В настоящем стандарте использованы нормативные ссылки на следующие стандарты [для датированных ссылок применяют только указанное издание ссылочного стандарта, для недатированных - последнее (включая все изменения)]:
ISO 11135, Sterilization of health-care products - Ethylene oxide - Requirements for the development, validation and routine control of a sterilization process for medical devices (Стерилизация медицинской продукции. Этиленоксид. Требования к разработке, валидации и текущему управлению процессом стерилизации медицинских изделий)
ISO 11137-1, Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices (Стерилизация медицинской продукции. Радиационная стерилизация. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий)
ISO 11139, Sterilization of health care products - Vocabulary - Terms used in sterilization and related equipment and process standards (Стерилизация медицинской продукции. Словарь терминов, используемых в стандартах на стерилизационное и аналогичное оборудование и процессы стерилизации)
ISO 13408-1:2008 <1>, Aseptic processing of health care products - Part 1: General requirements (Асептическое производство медицинской продукции. Часть 1. Общие требования)
--------------------------------
<1> Заменен на ISO 13408-1:2023. Однако для однозначного соблюдения требования настоящего стандарта, выраженного в датированной ссылке, рекомендуется использовать только указанное в этой ссылке издание.
ISO 13408-1:2008/Amd.1:2013 <1>, Aseptic processing of health care products - Part 1: General requirements - Amendment 1 (Асептическое производство медицинской продукции. Часть 1. Общие требования. Изменение 1)
--------------------------------
<1> Заменен на ISO 13408-1:2023. Однако для однозначного соблюдения требования настоящего стандарта, выраженного в датированной ссылке, рекомендуется использовать только указанное в этой ссылке издание.
ISO 13408-5, Aseptic processing of health care products - Part 5: Sterilization in place (Асептическое производство медицинской продукции. Часть 5. Стерилизация на месте)
ISO 13485, Medical devices - Quality management systems - Requirements for regulatory purposes (Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования)
ISO 17665-1, Sterilization of health care products - Moist heat - Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices (Стерилизация медицинской продукции. Влажное тепло. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий)
В настоящем стандарте применены термины по ИСО 11139, а также следующие термины с соответствующими определениями.
ИСО и МЭК поддерживают терминологические базы данных, используемые в стандартизации по следующим адресам:
- платформа онлайн-просмотра ИСО: доступна по адресу https://www.iso.org/obp;
- Электропедия МЭК: доступна по адресу http://www.electropedia.org/.
3.1 испытание на задерживание бактерий <*> (bacterial challenge test): Техническая операция, выполняемая для оценки способности фильтра (3.5) задерживать микроорганизмы из фильтруемой тестовой среды, содержащей бактерии, при установленных условиях.
--------------------------------
3.2 биологическая нагрузка (бионагрузка) <**> (bioburden): Популяция жизнеспособных микроорганизмов (3.9) на поверхности продукта или в продукте и/или стерильной барьерной системе.
Примечание 1 - В настоящем стандарте для термина "бионагрузка" также применимо определение - популяция жизнеспособных микроорганизмов в фильтруемой среде (3.6) до стерилизующей фильтрации (3.11).
--------------------------------
3.3 химическая совместимость <фильтра> (chemical compatibility): Возможность совместного использования обрабатываемых фильтруемых сред (3.6) и материалов фильтра (3.5) при заданных условиях процесса без негативного воздействия одного на другое.
3.4 экстрагируемое вещество (extractable): Вещество, выделяемое фильтром (3.5) или его материалом при использовании экстракционного раствора и/или в условиях экстракции по меньшей мере такой же химической активности, как при предполагаемом применении.
[ISO 10993-12:2012, 3.8, изменено - изменена формулировка].
3.5 фильтр (filter): Конструкция из пористого материала, через которую пропускают фильтруемую среду (3.6) для удаления жизнеспособных и/или нежизнеспособных частиц.
3.6 фильтруемая среда (fluid): Вещество (или смесь веществ), которое(ые) непрерывно деформируется(ются) (течет) под действием приложенной сдвигающей силы.
Пример - Жидкость, газ, пар или плазма.
Примечание 1 - Фильтрат фильтруемой среды, подвергаемой стерилизующей фильтрации (3.11), может являться производимой медицинской продукцией или ее компонентом, газом, используемым для обеспечения избыточного давления, или техническим газом, поступающим в зону асептической обработки (например, газы, поступающие из пневматических клапанов).
3.7 испытание фильтра на целостность (filter integrity test): Неразрушающее физическое испытание, которое подтверждает способность фильтра в собранном состоянии задерживать бактерии.
3.8 вымываемое вещество (leachable): Вещество, выделяемое фильтром (3.5) или компонентами собранного фильтра в условиях предполагаемого применения.
3.9 микроорганизм (microorganism): Микроскопические организмы, такие как бактерии, грибы, простейшие и вирусы.
Примечание 1 - В настоящем стандарте вирусы не рассматриваются.
3.10 номинальный размер пор (pore size rating): Заявленный и установленный на этикетке номинальный размер пор фильтра (3.5).
Примечание 1 - Номинальный размер пор определяется на основании показателей задерживания тестовых частиц. Номинальный размер пор не обязательно является физическим диаметром пор, это показатель, основанный на размере частиц, которые удаляются фильтром.
3.11 стерилизующая фильтрация (sterilizing filtration): Удаление жизнеспособных микроорганизмов (3.9) из фильтруемой среды (3.6) путем пропускания этой среды через фильтр (3.5) при заданных условиях процесса для получения стерильного фильтрата.
4.1 Общие положения
Для обеспечения надлежащего контроля всех выполняемых операций, влияющих на стерилизующую фильтрацию, должна быть внедрена система менеджмента качества, соответствующая ИСО 13408-1:2008, раздел 4, и ИСО 13408-1:2008/Amd.1:2013. Кроме того, должны соблюдаться требования, приведенные в 4.2 и 4.3 настоящего стандарта.
Необходимо обеспечить проведение и документирование обучения операторов, участвующих в процессе стерилизующей фильтрации, включающего изучение:
a) процедур фильтрации, видов отказов и необходимых предупреждающих мероприятий;
b) теории и практики по проведению испытаний на целостность;
c) процедур расследования отказов и мер, принимаемых в случае отклонений при проведении испытания на целостность;
d) процедуру сборки фильтра (включая асептическую технику, если необходимо);
e) процедур установки, очистки и стерилизации фильтра.
4.3.1 Необходимо установить процедуры закупки фильтров и фильтровального оборудования. Данные процедуры должны соответствовать применимым положениям ИСО 13485 или эквивалентной системы качества <*>.
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
4.3.2 Между пользователем фильтра и его производителем должно быть заключено письменное соглашение о том, что производитель будет уведомлять пользователя о любых изменениях условий производства фильтра, которые потенциально могут повлиять на определенные параметры фильтруемой среды и самого процесса.
4.3.3 Необходимо установить процедуры идентификации и прослеживаемости фильтров. Данные процедуры должны соответствовать применимым положениям стандарта ИСО 13485 или эквивалентной системы качества.
5.1 Общие положения
Разработка спецификации стерилизующего фильтра - это определение того, какие фильтры могут быть пригодны для использования в качестве стерилизующего фильтра в процессе стерилизующей фильтрации для данной фильтруемой среды. Обычно этот процесс выполняет пользователь фильтра с учетом информации, полученной от производителя фильтра.
Стерилизующие фильтры включают в том числе следующие виды:
a) мембранные фильтрующие диски, которые пользователь устанавливает в корпусы/держатели фильтров;
b) картриджные фильтры, которые пользователь устанавливает в корпусы/держатели фильтров;
c) устройства, поставляемые производителем фильтров в предварительно собранном виде (капсулы/патроны).
Спецификации для фильтров, используемых в производстве, должны соответствовать спецификациям для фильтров, используемых при валидации продукции и процесса фильтрации.
5.2 Эффективность удаления микроорганизмов
5.2.1 Для каждого сочетания стерилизующего фильтра и типа фильтруемой среды необходимо регистрировать данные об эффективности удаления микроорганизмов. Как правило, это делают при помощи испытания на задерживание с использованием:
a) взвеси микроорганизмов, специфичных для производимой продукции, при фильтрации жидкости и
b) типовых микроорганизмов в аэрозольной взвеси при фильтрации газов.
5.2.2 Необходимо определить характеристики процесса и их взаимодействие в отношении эффективности удаления микроорганизмов. Такими характеристиками в том числе являются:
a) характеристики фильтрующей мембраны, такие как распределение пор по размерам, химический состав поверхности, строение и тип полимера мембраны (см. 8.2.1);
b) характеристики фильтровального оборудования (см. 6.4);
c) характеристики фильтруемой среды, такие как влияние поверхностно-активных веществ или добавок, включая абсорбирующее влияние фильтруемой среды на микроорганизмы, pH, вязкость, осмолярность, поверхностное натяжение и ионная сила (см. 7.1.2);
d) бионагрузка на фильтруемую среду; количество, тип и размер клеток организмов, присутствующих в фильтруемой среде, а также технологические условия или рецептуры, которые могут повлиять на размер клеток (см. 7.1.2);
e) технологические условия, такие как размер серии, температура, перепад давления, скорость потока, время хранения и время фильтрации (см. 8.3.1);
f) влияние процесса стерилизации на эффективность фильтра.
В случае стерилизующей фильтрации газов некоторые из вышеперечисленных параметров могут быть неприменимы.
5.3 Влияние материалов
5.3.1 Необходимо оценить влияние экстрагируемых или вымываемых веществ из материалов фильтра(ов) на фильтруемые среды (см. 8.2.2.2 и 8.2.2.3).
5.3.2 Необходимо оценить влияние адсорбции продукции или ее компонентов на материал(ы) фильтра(ов) (см. 8.2.2.4).
5.3.3 Фильтры не должны выделять волокна.
Примечание - В качестве волокна, как правило, рассматривают частицу, имеющую соотношение сторон (отношение длины к ширине) 10 или более.
5.3.4 При повторном использовании фильтров проведение таких процессов, как разборка, очистка, ополаскивание, хранение, повторная сборка, промывка и стерилизация, должно быть обосновано. Необходимо оценить влияние этих процессов на эффективность удаления микроорганизмов и материалы фильтра (для более подробной информации см. 8.2.3.2).
5.4 Экологические аспекты
Процедуры уничтожения использованных материалов фильтра должны учитывать наличие в них фильтруемых веществ и обеспечивать безопасную утилизацию.
Примечание - Следует применять соответствующие требования по утилизации отходов.
6.1 Общие положения
Настоящий раздел содержит положения, обеспечивающие эффективность и воспроизводимость всего процесса стерилизующей фильтрации.
6.2 Управление рисками
6.2.1 При управлении рисками необходимо следовать ИСО 13408-1:2008, 5.2 и ИСО 13408-1:2008/Amd.1:2013 <*>, а также следующим требованиям.
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77 (см. часть 3).
6.2.2 При выборе фильтра и фильтровального оборудования проводят оценку рисков. Оценке подлежат в том числе, но не ограничиваясь этим, следующие риски:
a) влияние характеристик, указанных в 5.2.2;
b) конструкция стерилизующей фильтрационной системы в части включения и расположения в системе: фильтров для задерживания механических частиц или снижения биологической нагрузки, одиночных или последовательно расположенных стерилизующих фильтров, дополнительных или параллельных стерилизующих фильтров;
c) риск для стерильности системы фильтрации при проведении испытания фильтра на целостность перед использованием и после стерилизации (PUPSIT) <**>;
d) риски, связанные с повторным использованием фильтра для процесса стерилизующей фильтрации конкретной фильтруемой среды.
--------------------------------
6.2.3 Управление рисками включает оценку и управление рисками, связанными с передачей на аутсорсинг стерилизации критически стерильных компонентов, например фильтров, которые поставляются стерильными.
Для одноразовых систем фильтрации и фильтров управление рисками должно включать, но не ограничиваться этим, оценку следующих параметров:
a) конструкции системы фильтрации поставщика (включая потребность пользователя фильтра в конструкции из одиночного фильтра, последовательных, дополнительных или параллельных фильтров), используемых материалов, процессов производства и стерилизации;
b) расположения фильтра, т.е. внутри или снаружи изолятора;
c) технической возможности проведения испытания на целостность перед использованием и после стерилизации;
d) функционирования фильтрующей системы/фильтра, включая требования к промывке или смачиванию;
e) обеспечения стерильности после прохождения фильтра;
f) результатов проверки целостности закрытых систем;
g) воздействия собранного фильтра на отфильтрованную среду.
6.2.4 Оценку уровня риска и проверку эффективности процедур по снижению риска, как правило, проводят с использованием количественных показателей. Эти показатели должны включать результаты микробиологического мониторинга и мониторинга механических частиц в фильтруемой среде.
6.2.5 Результаты оценки рисков используют при разработке валидационного плана процесса стерилизующей фильтрации.
6.2.6 Управление рисками проводят системно. Оценку рисков следует обновлять по мере необходимости, в том числе в случае изменений процесса стерилизующей фильтрации и по результатам продолжающейся верификации.
6.3 Требования к процессу стерилизующей фильтрации
6.3.1 Необходимо указать параметры процесса и их допустимые значения. Определение допустимых значений должно быть основано на информации о сочетании параметров процесса, обеспечивающих минимально допустимую эффективность удаления микроорганизмов. Процесс фильтрации при таких параметрах должен стабильно обеспечивать получение стерильного фильтрата, соответствующего спецификации.
Установление допустимых значений для технологических параметров должно быть основано на анализе характеристик процесса (см. раздел 8).
6.3.2 Необходимо определить средства контроля и мониторинга параметров процесса.
6.3.3 Для обеспечения эффективности процесса стерилизующей фильтрации необходимо документировать любую обработку фильтруемой среды перед стерилизующей фильтрацией (например, использование фильтра, снижающего бионагрузку).
6.3.4 После стерилизующей фильтрации последующую асептическую обработку стерильного фильтрата осуществляют в соответствии с ИСО 13408-1 <*>.
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
6.4.1 Необходимо разработать спецификацию на оборудование, обеспечивающее эффективное выполнение процесса в пределах заданных значений характеристик процесса.
6.4.2 Спецификация должна включать в том числе, но не ограничиваясь, описание физических характеристик оборудования и необходимых вспомогательных устройств, включая материалы конструкции.
6.4.3 Выбор компонентов для системы фильтрации, их соединение и расположение в системе фильтрации необходимо обосновать и задокументировать.
Компоненты системы фильтрации не должны выделять примеси в фильтруемую среду или иным образом влиять на ее качество. К таким компонентам относятся:
a) системы трубопроводов и их соединения;
b) клапаны;
c) датчики и/или другие приборы;
d) прокладки, уплотнительные кольца и/или уплотнители;
e) материалы фильтра.
6.4.4 При фильтрации газов следует избегать непреднамеренного смачивания фильтра или накопления конденсата в фильтровальном оборудовании.
6.4.5 Проектирование системы фильтрации должно осуществляться в соответствии со следующими принципами:
a) обеспечение работы в пределах валидируемых параметров процесса;
b) обеспечение стерильности фильтрата (см. ИСО 13408-1:2008, раздел 6, и ИСО 13408-1:2008/Amd.1:2013);
c) размещение стерилизующего фильтра основано на оценке рисков.
Для того, чтобы минимизировать риск повторной контаминации фильтрата, рекомендовано использование систем с минимальным количеством соединений и прокладок. Расположение стерилизующего фильтра как можно ближе к оборудованию для наполнения (месту фасовки) или системе подачи также может снизить риск повторной контаминации;
d) отсутствие неприемлемого риска загрязнения производственной среды при процессе стерилизующей фильтрации;
e) обеспечение возможности проведения процедур очистки по мере необходимости;
f) обеспечение возможности стерилизации материала фильтра и всех компонентов или поверхностей, контактирующих с фильтруемой средой после фильтрации.
Примечание - Подходы к проведению стерилизации могут включать:
1) стерилизацию системы фильтрации на месте,
2) стерилизацию собранного фильтра пользователем с последующей асептической сборкой системы фильтрации или
3) поставку стерилизованных компонентов утвержденными поставщиками;
g) обеспечение проведения испытания на целостность на месте в закрытой системе после стерилизации фильтра до его использования - если указана необходимость проведения испытания PUPSIT;
h) документирование оборудования для мониторинга и управления процессом фильтрации, включая характеристики и расположение датчиков, а также индикаторных и регистрирующих приборов;
i) документирование неисправностей фильтровального оборудования;
j) определение мер безопасности, включая меры по защите персонала и производственной среды.
6.4.6 Необходимо обеспечить в соответствии с действующей системой менеджмента качества документированное подтверждение того, что программное обеспечение, используемое для управления и/или мониторинга процесса, соответствует своему целевому назначению.
Примечание - Следует руководствоваться ИСО/МЭК 90003 <*>.
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77 (Приложение N 11).
7.1 Общие характеристики
7.1.1 Необходимо определять характеристики фильтруемой среды, подлежащей стерилизации. Данную деятельность следует рассматривать как часть оценки рисков для выпускаемой продукции (см. ИСО 13408-1).
Примечание - В рамках настоящего раздела термин "фильтруемая среда" используют взамен термина "продукция". Продукцию обычно рассматривают как фильтрат, прошедший стерилизацию, или готовую продукцию, прошедшую асептическую обработку. Таким образом, при определении характеристик фильтруемой среды указывают характеристики, которые влияют на процесс фильтрации или сохраняются в фильтрате.
7.1.2 Необходимо указать и поддерживать в определенных пределах (где это применимо) следующие характеристики фильтруемой среды, подлежащей стерилизации:
a) состав;
b) pH;
c) осмолярность;
d) ионная сила;
e) вязкость;
f) плотность;
g) поверхностное натяжение;
i) механические частицы.
Примечание - Это необходимо для контроля и обеспечения бионагрузки (микробиологической чистоты) фильтруемой среды перед стерилизующей фильтрацией значительно ниже валидированного уровня эффективности задерживания системы фильтрации.
7.1.3 Необходимо оценить риски, связанные с веществами, которые могут выделяться из фильтра в фильтруемую среду, включая экстрагируемые и вымываемые вещества, механические частицы и эндотоксины.
7.1.4 Необходимо учитывать возможное влияние состава фильтруемой среды или материалов фильтра на размер задерживаемых микроорганизмов, что способствует прохождению микроорганизмов через стерилизующий фильтр.
7.1.5 Необходимо подтвердить соответствие фильтрата установленным требованиям эффективности, качества и эксплуатационным характеристикам после выполнения процесса стерилизующей фильтрации при наиболее неблагоприятных технологических параметрах для фильтруемой среды.
7.1.6 Если допускается проведение нескольких циклов фильтрации, необходимо оценить влияние такой обработки на фильтруемую среду.
7.2.1 Необходимо разработать и поддерживать систему контроля и мониторинга бионагрузки стерилизуемой среды для обеспечения эффективности процесса стерилизующей фильтрации [см. 5.2.2 d), 7.1.2 h), 7.1.4, 7.2.2, 8.2.3.1, 8.2.3.2].
7.2.2 Методы определения бионагрузки (микробиологической чистоты) должны соответствовать своему назначению, быть валидированы и задокументированы.
Примечание - Некоторые руководства приведены в ИСО 11737-1 и статьях Фармакопеи; например, "Микробиологическое исследование нестерильных продуктов: Испытания на определение численности микроорганизмов" приведено в Европейской Фармакопее (Ph. Eur.), Фармакопее Японии (JP) и Фармакопее США (USP) <*>.
--------------------------------
<*> В Российской Федерации следует руководствоваться статьей 2.3.1.2 "Требования к микробиологической чистоте лекарственных препаратов, фармацевтических субстанций и вспомогательных веществ для их производства" Фармакопеи ЕАЭС или ОФС.1.2.4.0002.18 "Микробиологическая чистота" Государственной фармакопеи.
8.1 Общие положения
8.1.1 Целью настоящего раздела является разработка подробной спецификации процесса стерилизующей фильтрации, который будет использован при производстве определенной продукции (см. раздел 7). Разработка спецификации включает в себя:
a) выбор наиболее подходящего стерилизующего фильтра (см. раздел 5);
b) выбор параметров процесса, обеспечивающих получение стерильного фильтрата и не оказывающих влияния на безопасность, эффективность и качество продукта (см. 5.2.2);
c) выбор метода стерилизации системы фильтрации [см. 6.4.5 f)].
8.1.2 Используемый процесс фильтрации должен быть определен и задокументирован в письменной процедуре.
Пользователь должен задокументировать обоснование выбора размера и типа фильтров, используемых в системе фильтрации для конкретной фильтруемой среды и процесса фильтрации. Обоснование должно включать следующее, но не ограничиваться этим:
a) эффективную площадь поверхности фильтра, необходимую для требуемой скорости потока;
b) значение номинального размера пор фильтра;
c) температурную совместимость материалов фильтра с температурами процесса и/или стерилизации;
d) гидравлическую устойчивость, позволяющую выдержать перепад давления процесса;
e) конфигурацию фильтра (плоский или картриджный), дополнительные, последовательные, параллельные фильтры;
f) срок эксплуатации фильтра;
g) бионагрузку на фильтруемую среду [см. 5.2.2 d)].
8.2.2 Совместимость фильтра и фильтруемой среды
8.2.2.1 Пользователь должен подтвердить совместимость фильтра и фильтруемой среды. Исследование совместимости должно включать, но не ограничиваться этим, изучение:
a) влияния состава фильтруемой среды и условий процесса на химические и физические свойства, а также эффективности фильтра;
b) влияния фильтра и условий процесса на соответствующие биологические, химические и физические показатели фильтруемой среды.
Оценка должна включать определение экстрагируемых и вымываемых веществ, механических частиц и адсорбцию.
8.2.2.2 При определении вымываемых веществ идентификацию и количественное определение материала, вымываемого из фильтра, проводят с использованием фильтруемой среды и тем же типом фильтра, который будет использоваться в производстве. Если невозможно использовать фильтруемую среду, может быть использована имитационная среда. Фильтруемые среды со схожими свойствами могут быть сгруппированы, а для испытания выбирают условия наихудшего случая. При использовании имитационной среды или группировке сред необходимо документированное обоснование.
Примечание - Промывка фильтров перед использованием может снизить уровень потенциально вымываемых веществ.
8.2.2.3 При оценке экстрагируемых веществ необходимо провести токсикологические исследования, подтверждающие отсутствие какой-либо значимой токсичности веществ, экстрагируемых из фильтра.
Примечание - Данные по экстрагируемым веществам и таблицы химической совместимости, как правило, предоставляются производителем фильтров и обычно используются в качестве отправной точки для определения необходимости дальнейших испытаний.
8.2.2.4 Необходимо оценить влияние адсорбции компонентов фильтруемой среды (на материале фильтра) на состав и концентрацию фильтрата.
Примечание - Адсорбция - это механизм связывания компонентов фильтруемой среды с материалом фильтра, что может повлиять на состав фильтрата. На адсорбцию могут влиять, например, скорость потока, концентрация компонентов, время экспозиции, температура и pH.
8.2.3 Использование фильтра
8.2.3.1 Если перед стерилизующей фильтрацией необходимо снизить бионагрузку, следует рассмотреть возможность использования фильтра предварительной очистки перед стерилизующим фильтром.
Примечание - К фильтрам предварительной очистки могут быть применены требования 8.2.2.1 - 8.2.2.3 с учетом их целевого назначения.
8.2.3.2 К стерилизующим фильтрам, предназначенным для повторного использования, предъявляются требования, указанные в 8.2, 8.3 и разделе 9.
Если стерилизующий фильтр предназначен для повторного использования, пользователь должен:
ИС МЕГАНОРМ: примечание. В официальном тексте документа, видимо, допущена опечатка: подпункт d) в пункте 6.2.1 отсутствует. |
a) провести оценку и задокументировать риски (для качества), связанные с повторным использованием фильтра в процессе стерилизующей фильтрации для данной фильтруемой среды [см. 6.2.1 d)];
b) провести и задокументировать надлежащие валидационные и квалификационные исследования для подтверждения того, что повторное использование фильтра для данного процесса стерилизующей фильтрации и для данной фильтруемой среды не ухудшает эффективность стерилизующего фильтра или качество фильтрата (см. раздел 9);
c) задокументировать валидированное и разрешенное для данного фильтра максимальное количество циклов повторного использования и внедрить меры контроля для обеспечения исключения повторного использования фильтров сверх утвержденного максимального количества циклов; необходимо вести записи о выполняемых мерах контроля;
d) внедрить меры контроля для обеспечения исключения использования для производства последующих серий продукта фильтров, контаминированных фильтруемой средой/остаточным количеством моющего раствора или признанных дефектными по иным причинам; необходимо вести записи о выполняемых мерах контроля.
8.3.1 Необходимо разработать, количественно охарактеризовать и документировать параметры процесса, включая следующее, но не ограничиваясь этим:
a) мониторинг таких характеристик, как температура, давление, скорость потока, общий объем и продолжительность процесса, для обеспечения того, чтобы параметры процесса оставались в пределах установленных допустимых значений;
b) процедуры стерилизации для собранного фильтра, системы фильтрации и системы транспортирования и резервуаров для фильтруемой среды, включая допустимый предел суммарного времени стерилизации и/или количества циклов при соответствующих условиях стерилизации в случае многократной стерилизации и повторного использования (см. 8.2.3.2);
c) конфигурацию и монтаж фильтра;
1) время хранения фильтруемой среды перед стерилизующей фильтрацией и влияние на бионагрузку;
2) подготовку (выдержку) фильтра в фильтруемой среде, при необходимости;
3) промывку фильтра фильтруемой средой;
4) время фильтрации/общее время экспозиции фильтра в фильтруемой среде;
5) максимальное количество повторных использований для стерилизующих фильтров;
6) скорость потока;
7) фильтруемый объем;
8) температуру;
9) перепад давления;
e) процедуры очистки системы фильтрации после использования.
Примечание 1 - При разработке и валидации процедуры очистки может быть использована информация об операции промывки от производителя фильтра.
Примечание 2 - При определении минимального объема жидкости для промывки могут быть использованы результаты испытания на определение общего органического углерода (TOC) или протеин-специфическое количественное определение.
8.3.2 Документированные процедуры должны содержать следующее, но не ограничиваться этим:
a) проверку компонентов системы фильтрации;
b) порядок сборки системы фильтрации;
c) очистку, стерилизацию и/или промывку;
d) период времени между очисткой и стерилизацией (если применимо);
e) период времени между стерилизацией и использованием;
f) контрольные испытания, включая испытание на целостность фильтра;
g) мониторинг параметров температуры, перепада давления, скорости потока;
h) период времени между фильтрацией среды и очисткой и т.д.
8.3.3 Необходимо установить документированные процедуры испытания фильтра на целостность, включающие критерии приемлемости и действия при получении неудовлетворительных результатов, а также условия, при которых испытание фильтра на целостность может быть проведено повторно (см. 8.4).
8.3.4 Необходимо обеспечить наличие процедур, минимизирующих количество микроорганизмов в фильтруемой среде перед стерилизующей фильтрацией, таким образом максимально снижая нагрузку на стерилизующий фильтр.
8.4.1 Стерилизующий фильтр, используемый для стерилизации фильтруемой среды, должен быть подвержен неразрушающему испытанию на целостность после использования, без извлечения фильтра из корпуса. Результаты испытания должны коррелировать с подтвержденной в процессе валидации способностью фильтра удерживать микроорганизмы.
8.4.2 Целостность системы фильтрации перед использованием имеет критическое значение для обеспечения стерильности продукта. Если повреждение фильтров, например во время транспортирования или стерилизации, или неправильная установка картриджных фильтров в корпусы не будут выявлены, это приведет к браку продукции в случае неудовлетворительного результата испытания на целостность после использования. Поэтому следует рассмотреть возможность проведения испытания на целостность до и после фильтрации. Решение об отказе от проведения испытания перед использованием фильтра должно быть основано на результатах оценки рисков для качества и быть задокументировано.
При проведении испытания на целостность стерильного фильтра перед использованием необходимо учитывать следующее:
a) испытание на целостность не должно нарушать стерильность простерилизованного фильтра или последующего технологического процесса;
b) среда, используемая для испытания на целостность, должна быть совместима с фильтруемой средой.
8.4.3 Если процесс валидируют в виде последовательной или параллельной системы фильтрации для достижения стерильности данной фильтруемой среды, то эту систему рассматривают как единый стерилизующий блок и все стерилизующие фильтры системы должны выдерживать испытание на целостность после использования.
8.4.4 Если при использовании дополнительной (дублирующей) системы фильтрации основной фильтр не прошел испытание на целостность после использования, то необходимо провести оценку рисков для определения приемлемости результатов испытания на целостность дополнительного (дублирующего) фильтра после использования. Оценку рисков проводят как часть процесса испытания на целостность, а не в случае получения неудовлетворительных результатов. Если при использовании дополнительной (дублирующей) системы фильтрации основной фильтр прошел испытание на целостность после использования, проведение испытания на целостность дополнительного (дублирующего) фильтра после использования не обязательно.
8.4.5 Если фильтры для газов используют в течение длительного времени, например "дыхательные" фильтры, то испытания на целостность проводят до и после использования. Продолжительность использования необходимо устанавливать в зависимости от времени простоя, времени (продолжительности) использования, количества операций (стерилизации), потока фильтруемой среды (объема, скорости) и т.д.
8.4.6 Необходимо установить документированные процедуры испытания на целостность фильтра, включая критерии приемлемости и действия при получении неудовлетворительных результатов, а также условия, при которых испытание может быть проведено повторно.
9.1 Общие положения
Целью валидации является подтверждение того, что процесс фильтрации, разработанный и описанный в соответствии с 8.3, обеспечивает эффективное и воспроизводимое получение стерильного фильтрата. Валидация включает в себя несколько последовательных этапов.
a) способность фильтра задерживать микроорганизмы в зависимости от характеристик фильтруемой среды (т.е. стерилизующая способность фильтрующего материала);
b) определение параметров испытания на целостность фильтра, специфичных для фильтруемой среды [согласно 9.1 a)];
c) взаимодействие с фильтруемой средой;
d) стерилизацию системы фильтрации.
Для валидации стерилизации газов (см. 9.6) оценивают:
e) подтверждение способности фильтра задерживать типовые микроорганизмы в аэрозольной взвеси в заданных условиях;
f) определение параметров испытания на целостность фильтров;
g) стерилизацию системы фильтрации.
9.2 Валидация эффективности задерживания микроорганизмов, специфичных для фильтруемой среды, стерилизующими фильтрами для жидкостей
9.2.1 Общие положения
9.2.1.1 Валидацию стерилизующей фильтрации жидкостей начинают с квалификации фильтра путем проведения испытания на задерживание соответствующих бактерий с использованием, по крайней мере, одного фильтра из не менее чем трех серий фильтров с получением трех последовательных успешных результатов для каждого испытания. Все неудовлетворительные результаты должны быть расследованы.
Примечание 1 - Эти испытания, как правило, проводят в лабораторных условиях с использованием уменьшенной модели системы фильтрации (которая может состоять из картриджа или диска другого размера с тем же фильтрующим материалом), чтобы не нарушить стерильность производственной среды.
Примечание 2 - Информация от производителя фильтра может быть использована при разработке и валидации испытаний на целостность фильтра.
Примечание 3 - Как правило, производители фильтров публикуют методики и результаты испытаний, обосновывающие промышленное применение фильтров для стерилизующей фильтрации. Эти документы могут быть использованы, но не могут заменять проведение валидации процесса фильтрации жидкости, выполняемой пользователем фильтра.
9.2.1.2 Для моделирования наихудшего случая в отношении задерживающей способности микроорганизмов необходимо использовать в испытании как минимум одну серию фильтров (материалов фильтра) со значениями показателя физической целостности, соответствующими или близкими к допустимому значению, указанному производителем фильтра.
9.2.1.3 При выборе условий, моделирующих наихудшие условия процесса, учитывают параметры, описанные в 8.3.1 d).
9.2.1.4 Жидкости с подобными свойствами могут быть сгруппированы, а для исследования задерживания микроорганизмов из группы может быть выбрана одна фильтруемая среда, представляющая собой наихудший случай. Группировку жидкостей и выбор среды в качестве наихудшего случая необходимо документально обосновать.
9.2.1.5 В качестве среды для испытания на задерживание микроорганизмов следует выбирать жидкость, подлежащую фильтрации. Должна быть оценена жизнеспособность испытуемых микроорганизмов в фильтруемой среде в течение всего времени испытания в условиях наихудшего случая. В случае, если фильтруемая среда не может быть использована из-за антимикробных или иных свойств, используют имитационную среду или изменяют условия проведения имитационного испытания. Такие изменения условий могут включать:
a) модификацию среды, подлежащей фильтрации, например, при помощи уменьшения или удаления антимикробного компонента и/или регулирования уровня pH;
b) использование имитационной среды, которая должна как можно точнее воспроизводить состав среды, подлежащей фильтрации, включая следующие характеристики: pH, вязкость, ионную силу, осмолярность, поверхностную активность/натяжение, плотность и воздействие среды на типовые микроорганизмы;
c) сокращение времени экспозиции среды на микроорганизмы;
d) снижение температуры среды во время испытания после предварительной экспозиции фильтра в среде при температуре реального процесса стерилизующей фильтрации;
e) использование микроорганизмов, устойчивых к антимикробным свойствам среды или процесса;
f) экспозицию фильтра в среде в течение времени, равного времени контакта фильтра с фильтруемой средой во время процесса стерилизующей фильтрации, и последующее испытание с использованием модифицированной среды, как указано в 9.2.1.5 a) или b).
9.2.2 Типовые микроорганизмы (тест-штаммы микроорганизмов)
9.2.2.1 Необходимо определить реальную бионагрузку среды, которая будет подвергаться стерилизующей фильтрации (см. также 7.2).
9.2.2.2 Если фильтруемая среда содержит микроорганизмы размером не менее размеров Brevundimonas diminuta, то в качестве типового микроорганизма для стерилизующего фильтра с размером пор 0,2 мкм следует выбирать Brevundimonas diminuta (т.е. ATCC 19146 <*>).
--------------------------------
9.2.2.3 Если предполагаемая бионагрузка может состоять из микроорганизмов размерами меньше, чем Brevundimonas diminuta, что не позволит оценить задерживающую способность стерилизующего фильтра, то выбирают микроорганизмы с соответствующими размерами (отличные от B. diminuta). Условия культивации должны обеспечивать получение микроорганизмов приемлемого размера. Необходимо оценить влияние фильтруемой среды на размер микроорганизмов.
Примечание - Факторы риска, влияющие на задерживающую способность фильтра, могут включать присутствие:
a) вещества, способного влиять на прохождение микроорганизма через мембрану фильтра (например, липосомы);
b) микроорганизмов, проходящих через фильтр;
c) плеоморфных организмов (например, L-форм в растворе пенициллина, микоплазмы, лептоспиры).
9.2.2.4 Если использование тест-штамма Brevundimonas diminuta невозможно, а микроорганизмы, способные проходить через фильтр, не были выявлены, пользователь фильтра должен обосновать выбор альтернативного микроорганизма. Если в качестве типовых микроорганизмов культивируют альтернативные микроорганизмы, условия культивации должны быть подобраны таким образом, чтобы получить микроорганизмы приемлемого размера.
9.2.2.5 Минимальное количество микроорганизмов для испытания должно составлять 1 x 107 колониеобразующих единиц на квадратный сантиметр (КОЕ/см2) эффективной площади поверхности фильтра.
9.2.2.6 Если для задерживания микоплазмы или других микроорганизмов размером менее размеров Brevundimonas diminuta выбран стерилизующий фильтр с размером пор 0,1 мкм, то в качестве типового микроорганизма может быть использован Acholeplasma laidlawii (ATCC 23206) или аналогичные микроорганизмы [9]. При этом минимальное количество микроорганизмов для испытания должно составлять 1 x 107 колониеобразующих единиц на квадратный сантиметр (КОЕ/см2) эффективной площади поверхности фильтра.
В случае, если стерилизующий фильтр с размером пор 0,1 мкм используют только для удаления микроорганизмов, размер которых равен или превышает размер Brevundimonas diminuta, см. 9.2.2.2 - 9.2.2.5.
9.2.2.7 Валидация микробиологических методик, используемых для испытания на задерживание микроорганизмов, должна обеспечивать следующее:
a) микроорганизмы для испытания диспергируются в таком объеме фильтруемой среды, чтобы общий объем фильтрата был репрезентативен размеру производственной серии и эффективной площади фильтрации, за исключением случаев, когда антимикробные свойства требуют иного подхода.
Примечание 1 - При проведении испытания на задерживание бактерий с использованием уменьшенной модели системы фильтрации может потребоваться рециркуляция фильтруемой среды;
b) оценку жизнеспособности используемых микроорганизмов проводят на основании соответствующего количества образцов, отобранных в течение всего времени проведения испытания, чтобы доказать, что ожидаемая бактериальная нагрузка достигнута и остается стабильной в течение всего времени проведения испытания;
c) испытание на задерживание микроорганизмов проводят в условиях наихудшего случая, возможного при обычной работе;
d) используемый для испытания микроорганизм имеет минимальный размер.
Примечание 2 - Это может быть достигнуто при помощи использования положительного контроля для подтверждения прохождения типового микроорганизма через фильтр (например, прохождение B. diminuta через мембрану с размером пор 0,45 мкм или прохождение A. laidlawii через мембрану с размером пор 0,2 мкм);
e) используемая аналитическая методика позволяет выявлять небольшое количество микроорганизмов.
9.2.2.8 Критерии приемлемости должны включать требование о задержании всех микроорганизмов тремя испытуемыми фильтрами. В каждом испытании должен выполняться положительный контроль.
9.2.2.9 Данные об эффективности фильтра, полученные в результате испытания на задерживание микроорганизмов, используют для определения минимальных требований к проведению испытания на целостность фильтра, используемого на производстве.
Примечание 1 - Если установлена воспроизводимая взаимосвязь между способностью стерилизующего фильтра задерживать микроорганизмы, специфичные для фильтруемой среды, и физической целостностью данного фильтра, то используют подходящие неразрушающие испытания на целостность фильтра до и после его использования, чтобы определить, успешно ли прошел процесс фильтрации произведенной серии продукции (см. 8.4).
Примечание 2 - Настоящие требования используют для определения успешности проведенного процесса фильтрации производимой продукции, так как испытание на задерживание бактерий является разрушающим испытанием, которое нецелесообразно проводить для производственных фильтров.
9.2.2.10 Пользователь фильтра должен установить рабочие условия процесса стерилизующей фильтрации, которые должны будут находиться в пределах значений параметров, подтвержденных в процессе валидации с получением стерильного фильтрата.
9.3 Валидация методики испытания на целостность стерилизующих фильтров для жидкостей
Смачивающую жидкость для испытания на целостность фильтра выбирают либо из перечня рекомендованных производителем фильтра стандартных смачивающих жидкостей (например, вода, раствор изопропилового спирта), либо из жидкостей, подлежащих фильтрации (например, лекарственный препарат, промежуточные продукты и буферные растворы). При выборе последнего варианта необходимо определить и обосновать соответствующие спецификации.
Фильтруемую среду следует использовать для испытания на целостность фильтра в тех случаях, когда:
a) стандартная смачивающая жидкость, остающаяся на фильтре после испытания, оказывает неблагоприятное воздействие на фильтруемую среду или процесс фильтрации;
b) использование стандартной смачивающей жидкости может представлять риск для стерильности процесса фильтрации;
c) для удаления среды из фильтра требуется длительная операция промывки стандартной смачивающей жидкостью.
9.4 Валидация взаимодействия фильтра с фильтруемой средой
Необходимо провести определение следующих веществ:
a) вымываемые вещества (см. 8.2.2.2);
b) экстрагируемые вещества (см. 8.2.2.3);
c) материалы, удаляемые из фильтруемой среды путем абсорбции (см. 8.2.2.4).
9.5.1 Пользователь фильтра должен документально подтвердить, что:
a) процесс стерилизации сохраняет эффективность при минимальных условиях воздействия;
b) максимальные условия воздействия не оказывают негативного влияния на систему фильтрации.
9.5.2 Процессы стерилизации валидируют в соответствии с положениями ИСО 13408-5, ИСО 17665-1, ИСО 11135 или ИСО 11137-1 <*>.
Примечание - Типовыми процессами стерилизации систем фильтрации являются стерилизация паром на месте (SIP <**>) или автоклавирование, выполняемые пользователем, а также стерилизация оксидом этилена или радиационная стерилизация, проводимые производителем фильтров, если фильтры или системы фильтрации поставляются стерилизованными.
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77, положения Фармакопеи ЕАЭС и Государственной фармакопеи.
9.6 Валидация эффективности задерживания микроорганизмов, специфичных для фильтруемой среды, стерилизующими фильтрами для газов
9.6.1 Общие положения
Процесс стерилизующей фильтрации газов, как правило, валидируют при помощи аэрозольной взвеси типовых микроорганизмов. В настоящее время информация о влиянии газа-носителя на процесс стерилизующей фильтрации отсутствует. Поэтому проведение испытания на задерживание микроорганизмов, специфичных для процесса и фильтруемой среды, обычно не требуется.
Примечание - Как правило, производители фильтров публикуют методики и результаты испытаний, используемые для обоснования промышленного применения фильтров для стерилизующей фильтрации.
9.6.2 Задерживание аэрозоля микроорганизмов
Оценивают возможность применения данных о задерживании аэрозолей, предоставленных производителем фильтра, к используемому процессу стерилизующей фильтрации.
9.6.3 Валидация методики испытания на физическую целостность фильтра
9.6.3.1 Оценивают возможность применения данных, предоставленных производителем фильтров, к используемому процессу стерилизующей фильтрации.
9.6.3.2 Результаты испытания на физическую целостность фильтра должны коррелировать с его эффективностью задерживания микроорганизмов.
Примечание - Производителям фильтров следует представлять данные об испытании на целостность в своих руководствах по валидации.
9.6.3.3 Для испытания гидрофобных фильтрующих материалов можно применять традиционные подходы, используемые для гидрофильных фильтрующих элементов. Смачивающую жидкость для испытания на целостность фильтра выбирают из перечня рекомендованных производителем фильтра стандартных смачивающих жидкостей (например, раствор изопропилового или трет-бутилового спирта). Необходимо указать используемые при испытании смачивающую жидкость и газ, а испытание проводить при температурах, рекомендованных производителем фильтра. В качестве альтернативного метода возможно проведение испытания на проникновение воды. В некоторых случаях при проведении испытания на целостность фильтра возможно применение аэрозоля.
9.6.4 Совместимость и срок эксплуатации
Совместимость фильтра в реальных условиях эксплуатации подтверждают путем проведения испытания на целостность фильтра до и после экспозиции в условиях эксплуатации.
Необходимо регистрировать данные, подтверждающие целостность фильтра на протяжении всего срока эксплуатации.
Примечание - В большинстве случаев фильтры, прошедшие стерилизацию, используют непрерывно или многократно.
9.6.5 Валидация процесса стерилизации системы фильтрации для газов
См. 9.5.
10.1 Целью текущего мониторинга и контроля является подтверждение валидированного статуса <*> на протяжении всего процесса стерилизующей фильтрации.
--------------------------------
<*> Т.е. продолжающаяся верификация. Для производства лекарственных средств см. приложение N 15 "Правил надлежащей производственной практики Евразийского экономического союза", утвержденных Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
10.2 Необходимо подтверждать испытаниями то, что процесс стерилизующей фильтрации был проведен в пределах установленных допустимых значений.
10.3 Необходимо регистрировать данные, подтверждающие достижение параметров процесса в пределах установленных допустимых значений.
10.4 Все записи должны храниться в соответствии с ИСО 13485 или эквивалентной системой качества <**>.
--------------------------------
<**> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
10.5 Бионагрузку перед фильтрацией следует определять для каждой серии, за исключением случаев, когда документально подтверждено, что ее значение всегда находится в пределах допустимых значений.
10.6 При необходимости перед фильтрацией для каждой серии определяют уровень механических частиц, за исключением случаев, когда все параметры производства тщательно контролируются и результаты предыдущих испытаний подтверждают, что уровень механических частиц всегда находится в пределах допустимых значений.
10.7 Испытание на физическую целостность стерилизующего фильтра при помощи валидированной аналитической методики проводят после каждого использования без извлечения фильтра из корпуса.
11.1 Необходимо определить процедуру выпуска в обращение продукции, прошедшей стерилизующую фильтрацию. В данной процедуре должны быть установлены критерии, позволяющие подтвердить соответствие процесса стерилизующей фильтрации его спецификации.
11.2 Все параметры процесса фильтрации, признанные критическими, должны быть задокументированы. Эта документация должна быть частью записей по производству серии.
a) даты подготовки и фильтрации среды;
b) наименование и номер серии фильтруемой среды;
c) ФИО оператора(ов);
d) производитель фильтра, тип фильтра и номер(а) партии и/или серийный(ые) номер(а), присвоенные производителем фильтра;
e) данные об очистке системы фильтрации;
f) данные об условиях стерилизации системы фильтрации;
g) данные о циклах стерилизации, используемые для компонентов, применяемых в процессе фильтрации;
h) номер цикла (из установленного допустимого количества циклов) в случае повторного использования;
i) параметры процесса фильтрации (например, перепад давления, значение давления на входе и на выходе из фильтровальной системы, скорость потока, рабочая температура, время и т.д.);
j) результаты испытаний на целостность фильтра и их оценка;
k) результаты определения бионагрузки до начала процесса стерилизующей фильтрации;
l) отклонения от документированных процедур;
m) ФИО лица, ответственного за процесс стерилизующей фильтрации.
11.4 Если отсутствуют записи, указанные в 11.3, продукцию признают несоответствующей и обращаются с ней в соответствии с установленными процедурами (см. ИСО 13485 <*>).
--------------------------------
<*> В Российской Федерации при производстве лекарственных средств следует учитывать "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
12.1 Общие положения
Необходимо осуществлять продолжающуюся верификацию системы фильтрации для подтверждения эффективности стерилизующей фильтрации фильтруемой среды.
12.2 Повторная калибровка
Точность и надежность приборов, используемых для контроля, индикации или регистрации параметров процесса стерилизующей фильтрации, необходимо периодически проверять в соответствии с ИСО 13408-1:2008, 4.3.2, и ИСО 13408-1:2008/Amd.1:2013.
12.3 Техническое обслуживание оборудования
12.3.1 Профилактическое техническое обслуживание должно планироваться и выполняться в соответствии с документированными процедурами. Необходимо разработать порядок действий для каждого этапа, входящего в плановое техническое обслуживание, и определить частоту, с которой они должны выполняться. Записи о техническом обслуживании должны храниться в соответствии с ИСО 13408-1:2008, 4.1.4, и ИСО 13408-1:2008/Amd.1:2013.
12.3.2 До корректного завершения и регистрации всех этапов технического обслуживания оборудование не может быть допущено к эксплуатации.
12.3.3 Схема проведения, действия и записи о техническом обслуживании должна периодически оцениваться назначенным лицом. Результаты оценки должны регистрироваться в соответствии с ИСО 13408-1:2008, 4.1.4, и ИСО 13408-1:2008/Amd.1:2013.
12.4 Повторная квалификация (валидация)
12.4.1 Необходимо обосновать объем проводимой квалификации.
12.4.2 Архивные данные о процессе оценивают через определенные интервалы времени, чтобы определить необходимость повторной квалификации.
12.4.3 Оценку требований к повторной квалификации следует рассматривать как часть оценки конкретных изменений и последующего технического обслуживания.
12.4.4 Процедуры повторной квалификации должны быть определены, а записи о ней - сохранены.
12.4.5 Данные о повторной квалификации оценивают на соответствие установленным критериям приемлемости в соответствии с документированными процедурами. Записи о результатах оценки данных повторной квалификации хранят совместно с записями о внесенных исправлениях и предпринятых корректирующих действиях.
12.5 Оценка изменений
Любое изменение оценивают с точки зрения его влияния на эффективность процесса стерилизующей фильтрации. Оцениваемые изменения включают следующее, но не ограничиваются этим:
a) замену части системы фильтрации, которая может привести к изменению параметров процесса;
b) любое изменение состава фильтруемой среды;
c) любое изменение бионагрузки фильтруемой среды;
d) любые изменения стерилизующего фильтра;
e) любые изменения в условиях производства фильтра, о которых сообщает производитель фильтра и которые оценивают с точки зрения их потенциального влияния на определенные параметры фильтруемой среды и процесса (см. 4.3.2);
f) любое изменение параметров процесса;
g) новое или модифицированное программное или аппаратное обеспечение.
Необходимо документировать результаты оценки, включая обоснование принятых решений и объем изменений, внесенных в процесс стерилизующей фильтрации, или необходимость повторной квалификации.
(справочное)
Примечание - Для удобства применения нумерация пунктов в настоящем приложении соответствует нумерации пунктов основной части настоящего стандарта.
А.1 Общие положения
Руководство, приведенное в данном приложении, не является контрольным перечнем вопросов для проведения оценки соответствия настоящему стандарту, а предназначено для помощи в достижении однозначного понимания и применения настоящего стандарта путем предоставления разъяснений и приемлемых методов достижения соответствия установленным требованиям. Приложение содержит описание важных аспектов и примеры. Допускается использование методов, отличных от приведенных в настоящем руководстве, при условии, что их эффективность обеспечивает выполнение требования настоящего стандарта.
А.2 Примечание к нормативным ссылкам
См. раздел 2.
А.3 Примечание к терминам и определениям
Рисунок А.1 демонстрирует элементы стерилизующего фильтра и соответствующие им термины.
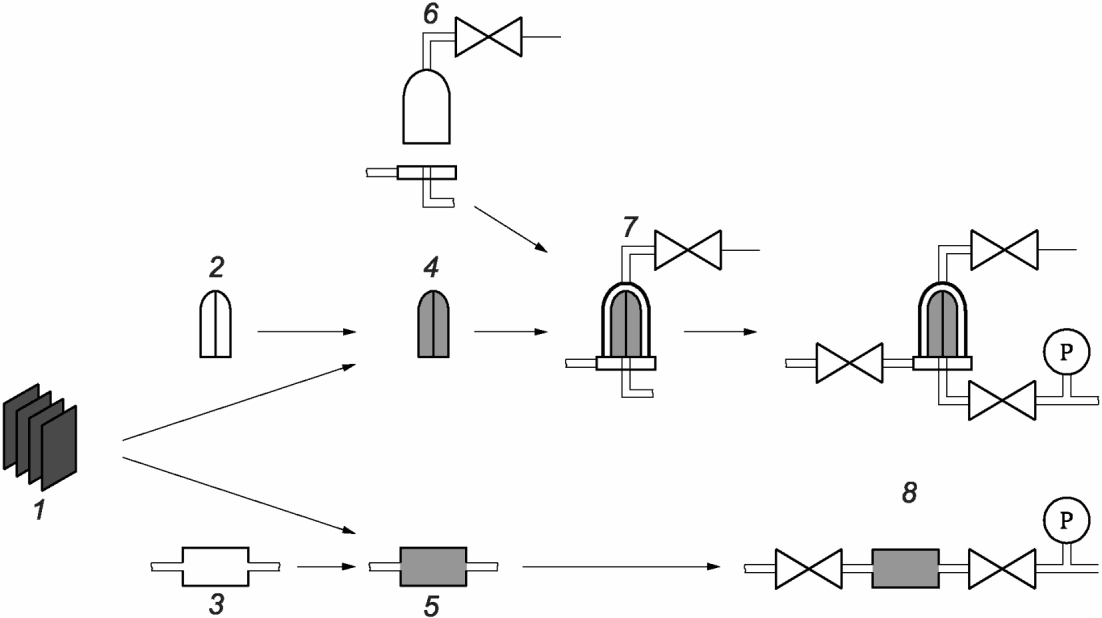
1 - материал фильтра; 2 - каркас картриджного фильтра;
3 - каркас капсульного фильтра; 4 - картриджный фильтр;
5 - капсульный фильтр (патрон); 6 - корпус фильтра;
7 - собранный фильтр; 8 - система фильтрации
Материал фильтра (1) представляет собой мембрану/матрицу, которая удаляет микроорганизмы из фильтруемой среды. Этот материал может быть помещен производителем фильтра в каркас (2), формируя картриджный фильтр (4). Затем картридж устанавливается пользователем в корпус фильтра (6) и стерилизуется. Картридж в корпусе часто называют собранным фильтром (7).
Также фильтрующий материал может быть помещен в капсульный каркас (3), формируя предварительно собранный капсульный фильтр (патрон) (5), который может поставляться пользователю в стерильном или нестерильном виде.
Собранный фильтр, соединенный с соответствующими клапанами, манометрами и т.д., называют системой фильтрации (8).
А.4 Элементы системы качества
А.4.1 Общие положения
Рекомендации отсутствуют.
А.4.2 Ответственность руководства
Рекомендации отсутствуют.
А.4.3 Поставка фильтров
Примечание - Настоящий пункт устанавливает требования к закупке фильтров и оборудования для фильтрации, используемых для реализации продукции. Требования к управлению закупками исходных материалов для производства продукции, подвергающейся стерилизующей фильтрации, приведены в ИСО 13408-1 <*>.
--------------------------------
<*> В Российской Федерации для производства лекарственных средств действуют "Правила надлежащей производственной практики Евразийского экономического союза", утвержденные Решением Совета ЕЭК от 3 ноября 2016 г. N 77.
А.4.3.1 Рекомендации отсутствуют.
А.4.3.2 Пользователь может проверить соблюдение письменных соглашений посредством аудита производителя фильтра.
А.4.3.3 Рекомендации отсутствуют.
А.5 Требования к стерилизующему фильтру
А.5.1 Общие положения
Обычно производитель фильтров предоставляет следующую основную информацию о фильтре:
e) рекомендуемая(ые) процедура(ы) стерилизации (общее время, количество циклов и условия стерилизации);
f) термостойкость;
g) способность задерживать механические частицы;
h) максимально допустимый перепад давления;
i) характеристики потока;
j) характеристики выделения частиц и/или волокон (миграция фильтрующего материала) в типовых растворителях (например, в воде);
k) эффективность задерживания микроорганизмов и ее корреляцию с результатами испытания на целостность фильтра при заданных условиях;
l) номинальный размер пор фильтра;
m) рекомендуемые методики испытания на целостность фильтра;
n) данные о биологической безопасности.
Сертификаты качества конкретных серий картриджных фильтров могут содержать следующую информацию, но не ограничиваться этим:
o) результаты испытания на целостность фильтра;
p) результаты испытания на бактериальные эндотоксины или пирогенность;
q) результаты испытаний на задерживание микроорганизмов;
r) наличие окисляемых веществ или общего органического углерода;
s) наличие экстрагируемых веществ;
t) характеристики выделяемых волокон и частиц;
u) данные о биологической безопасности;
v) значение скорости потока воды;
w) значение допустимых гидравлических нагрузок;
x) значение допустимых температур.
Примечание 1 - Пункты a), b), c) и d) обычно указывают на основании испытаний, проведенных для каждой серии.
Примечание 2 - Сертификаты качества обычно выдаются на картриджные фильтры, но могут быть также выданы на дисковые или пластинчатые фильтры.
А.5.2 Эффективность удаления микроорганизмов
Рекомендации отсутствуют.
А.5.3 Влияние материалов
А.5.3.1 Как правило, из фильтра вещества могут экстрагироваться или вымываться в жидкость. При фильтрации газов в исключительных случаях может потребоваться учет влияния экстрагируемых и вымываемых веществ.
А.5.3.2 Как правило, на фильтр вещества адсорбируются из жидкостей. В исключительных случаях может потребоваться учет адсорбции из газов.
А.5.3.3 Рекомендации отсутствуют.
А.5.3.4 Рекомендации отсутствуют.
А.5.4 Экологические аспекты
Рекомендации отсутствуют.
А.6 Требования к процессу и оборудованию
А.6.1 Общие положения
Рекомендации отсутствуют.
А.6.2 Управление рисками
А.6.2.1 Рекомендации отсутствуют.
А.6.2.2 Рекомендации приведены для 6.2.2 b).
Существует множество различных конфигураций асептических процессов с использованием систем стерилизующей фильтрации; ниже приведены примеры некоторых из них.
a) Процесс, в котором использован только один стерилизующий фильтр, как правило, с номинальным размером пор 0,2 мкм или менее (см. рисунок А.2).
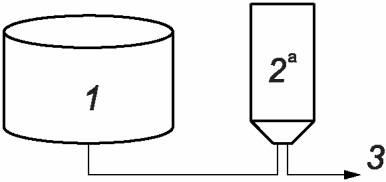
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - стерилизующий фильтр; 3 - стерильный фильтрат
стерилизующего фильтра
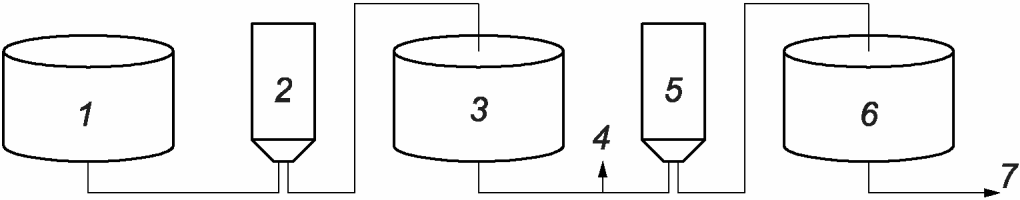
b) Процесс, в котором перед стерилизующим фильтром используют фильтр, снижающий количество частиц и/или бионагрузку (как правило, имеет номинальный размер пор 0,45 мкм или 0,2 мкм) (см. рисунок А.3). Данный процесс минимизирует и контролирует нагрузку на стерилизующий фильтр. Если бионагрузка нерасфасованной фильтруемой среды составляет более 10 КОЕ/100 мл, следует рассмотреть возможность использования системы фильтрации, приведенной ниже.
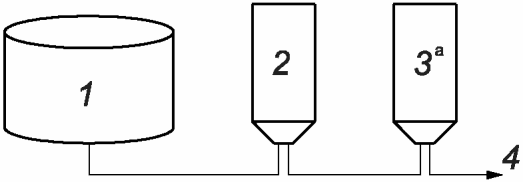
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильна);
2 - фильтр, снижающий бионагрузку; 3 - стерилизующий фильтр;
4 - стерильный фильтрат
снижающего бионагрузку, и стерилизующего фильтра
c) Процесс, в котором сначала используют фильтр, снижающий количество частиц и/или бионагрузку (как правило, с номинальным размером пор 0,45 мкм или 0,2 мкм), затем хранят продукт с низким уровнем бионагрузки, после чего используют стерилизующий фильтр (см. рисунок А.4). Данную систему фильтрации, как правило, используют в тех случаях, когда необходимо хранить фильтруемый раствор в течение ограниченного периода времени. Стерильность конечного фильтрата зависит от целостности стерилизующего фильтра.
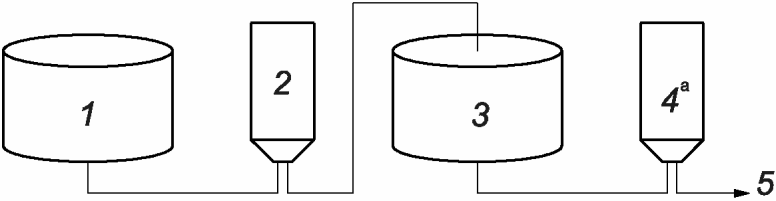
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - фильтр, снижающий бионагрузку; 3 - емкость для хранения
промежуточного продукта с низкой бионагрузкой;
4 - стерилизующий фильтр; 5 - стерильный фильтрат
снижающего количество частиц и/или бионагрузку
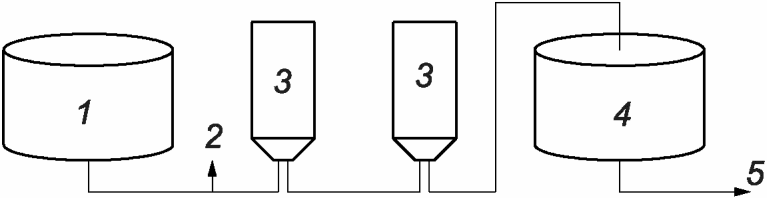
d) Процесс, в котором используют два или более последовательно расположенных стерилизующих фильтра (см. рисунок А.5). В данной системе среда последовательно фильтруется через два или более фильтров (расположенных один за другим) с одинаковым или последовательно уменьшающимся номинальным размером пор. В таком случае систему фильтров рассматривают как единый стерилизующий блок. Для обеспечения надлежащего валидированного процесса стерилизации оба фильтра должны сохранять целостность.
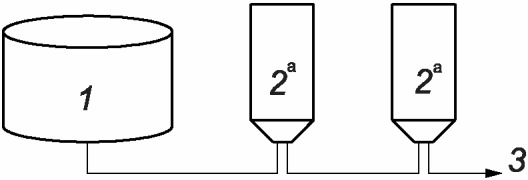
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - стерилизующий фильтр; 3 - стерильный фильтрат
последовательно установленных фильтров
e) Процесс, в котором используют идентичный стерилизующий фильтр, являющийся дополнительным (данную систему часто называют дополнительной системой фильтрации) (см. рисунок А.6). В данной системе эффективность задерживания микроорганизмов валидируется с использованием только одного из двух фильтров. Дополнительный фильтр является дублирующим и не нуждается в проверке на целостность после использования, за исключением случаев, когда результаты испытания на целостность основного фильтра были неудовлетворительными. Дополнительный фильтр может быть использован для принятия решения о качестве фильтрата в случае получения неудовлетворительных результатов испытания на целостность основного стерилизующего фильтра после использования.
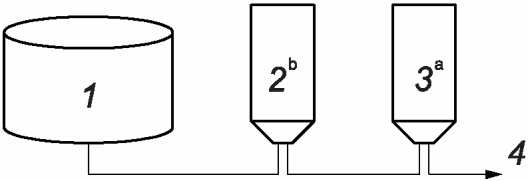
--------------------------------
<a> Необходимо испытание на целостность фильтра.
<b> Испытание на целостность фильтра необходимо только в случае получения неудовлетворительных результатов испытания на целостность основного фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - дополнительный стерилизующий фильтр; 3 - основной
стерилизующий фильтр; 4 - стерильный фильтрат
дополнительного стерилизующего фильтра
f) Процесс, включающий использование двух или более последовательно расположенных стерилизующих фильтров, а также этап промежуточного хранения продукта. Сначала продукт фильтруют через стерилизующий фильтр в стерильную емкость для хранения, затем продукт фильтруют через второй стерилизующий фильтр во время транспортирования/подачи на линию наполнения (см. рисунок А.7). Данную систему фильтрации, как правило, используют в тех случаях, когда при наполнении необходимо хранить раствор в течение определенного периода времени. Стерильность хранящегося промежуточного продукта и конечного фильтрата зависит от целостности обоих фильтров.
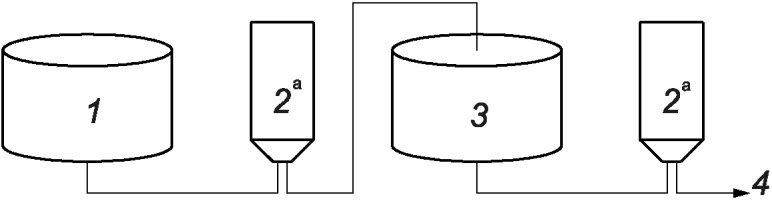
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - стерилизующий фильтр; 3 - емкость для хранения
промежуточного стерильного продукта; 4 - стерильный фильтрат
фильтрацию и хранение промежуточного стерильного продукта
g) Процесс, в котором используют параллельно соединенные стерилизующие фильтры, в результате чего технологический поток разделяется и равномерно фильтруется через несколько фильтров (см. рисунок А.8). Данную систему фильтрации, как правило, используют для увеличения производительности или скорости фильтруемого потока - все фильтры должны сохранять целостность.
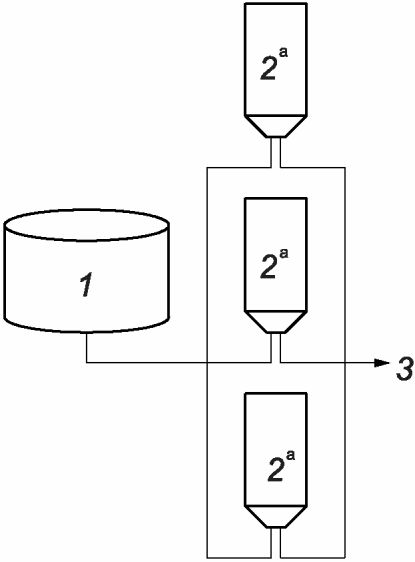
--------------------------------
<a> Необходимо испытание на целостность фильтра.
1 - нерасфасованная фильтруемая среда (нестерильная);
2 - стерилизующий фильтр; 3 - стерильный фильтрат
соединенных стерилизующих фильтров
Примеры часто встречаемых систем фильтрации приведены на рисунках А.9 и А.10.
1 - нестерильный раствор; 2 - фильтр, снижающий бионагрузку,
с номинальным размером пор 0,45 мкм; 3 - нерасфасованный
раствор с определенным уровнем бионагрузки; 4 - точка отбора
проб для контроля бионагрузки; 5 - стерилизующий фильтр
с номинальным размером пор 0,22 мкм; 6 - стерильный раствор;
7 - к линии наполнения
с использованием фильтра, снижающего бионагрузку,
вспомогательной емкости для хранения раствора с определенным
уровнем бионагрузки, точки отбора проб для контроля
бионагрузки и стерилизующего фильтра
1 - нестерильный раствор; 2 - точка отбора проб для контроля
бионагрузки; 3 - стерилизующий фильтр с номинальным размером
пор 0,22 мкм; 4 - стерильный раствор; 5 - к линии наполнения
последовательно соединенных фильтров для контроля
бионагрузки и механических частиц или дополнительной
(повторной) стерилизующей фильтрации
А.6.2.3 Рекомендации отсутствуют.
А.6.2.4 Рекомендации отсутствуют.
А.6.2.5 Рекомендации отсутствуют.
А.6.2.6 Рекомендации отсутствуют.
А.6.3 Требования к процессу стерилизующей фильтрации
Рекомендации отсутствуют.
А.6.4 Требования к оборудованию
А.6.4.1 Рекомендации отсутствуют.
А.6.4.2 Рекомендации отсутствуют.
А.6.4.3 Рекомендации отсутствуют.
А.6.4.4 Рекомендации отсутствуют.
А.6.4.5 Рекомендации приведены для 6.4.5 b).
В системе фильтрации с последовательными стерилизующими фильтрами чрезвычайно важно обеспечивать стерильность для всех операций между стерилизующими фильтрами, включая испытание на целостность фильтра перед использованием (если она проводится после стерилизации) и сам процесс фильтрации.
А.6.4.6 Рекомендации отсутствуют.
А.7 Определение характеристик фильтруемой среды
А.7.1 Общие характеристики
А.7.1.1 Рекомендации отсутствуют.
А.7.1.2 Важно обеспечивать однородность и воспроизводимость серий фильтруемой среды, подвергающейся стерилизации, в установленных пределах. Данное условие лежит в основе валидации стерилизующей фильтрации для конкретной фильтруемой среды. На основании полученных результатов устанавливаются подходящие значения скорости потока, времени процесса стерилизации и т.д. для текущего производственного цикла.
А.7.1.3 Рекомендации отсутствуют.
А.7.1.4 Рекомендации отсутствуют.
А.7.1.5 Рекомендации отсутствуют.
А.7.1.6 Рекомендации отсутствуют.
А.7.2 Микробиологическая чистота
А.7.2.1 Следует проанализировать данные о микробиологической чистоте исходных материалов и промежуточных продуктов (если применимо), а также для фильтруемой среды, подлежащей стерилизации, чтобы определить характеристики и уровень микробиологической чистоты, как правило, характерные для среды перед стерилизующей фильтрацией. Этот анализ должен включать следующее:
a) оценку того, имеют ли микроорганизмы, составляющие бионагрузку фильтруемой среды или исходных материалов, более малый размер, чем типовые микроорганизмы, используемые для определения эффективности стерилизующего фильтра задерживать микроорганизмы;
b) если размер микроорганизмов, составляющих бионагрузку, меньше, чем размер стандартных типовых микроорганизмов, используемых для определения эффективности стерилизующего фильтра задерживать микроорганизмы, проводят оценку риска прохождения микроорганизмов, составляющих бионагрузку, через один или несколько стерилизующих фильтров в контролируемых условиях эксплуатации.
Необходимо подтвердить эффективность системы контроля бионагрузки.
А.7.2.2 Рекомендации отсутствуют.
А.8 Определение характеристик процесса
А.8.1 Общие положения
Рекомендации отсутствуют.
А.8.2 Характеристики фильтра
А.8.2.1 Общие положения
Производители фильтров, как правило, публикуют результаты испытаний, проведенных в соответствии с применимыми фармакопейными методами для квалификации фильтра. Испытания, обычно проводимые производителем фильтров, могут включать:
a) задерживание бактерий в воде или питательной среде (лактозный бульон в растворе натрия хлорида) и корреляцию полученных результатов с результатами испытания на целостность фильтра;
b) химическую стойкость фильтра и ее влияние на целостность фильтра;
c) экстрагируемые вещества;
d) методы стерилизации и их влияние на целостность фильтра;
e) испытание на целостность фильтра в воде или растворителе;
f) испытание на токсичность;
g) испытание на механические частицы;
h) отсутствие выделения волокон материала;
i) общий органический углерод (TOC) и электропроводность.
А.8.2.2 Совместимость фильтра и фильтруемой среды
К факторам риска, влияющим на асептическую фильтрацию, относят неоднородность фильтра, химическое воздействие на фильтруемую среду, механические дефекты конфигурации, избыточное давление, нестерильность фильтровального оборудования и чрезмерную бионагрузку. Фильтры производят в различных размерах, конфигурациях и с различными химическими составами материалов фильтров.
А.8.2.3 Использование фильтра
А.8.2.3.1 Рекомендации отсутствуют.
А.8.2.3.2 Рекомендации приведены для 8.2.3.2.
Гидрофобные стерилизующие фильтры, используемые для стерилизующей фильтрации газов или в качестве "дыхательных" фильтров, часто применяют многократно; стерилизуют их паром между сериями продукции или производственными компаниями <*>.
--------------------------------
<*> Производство на основе компании - выделение производства одной продукции с несколькими сериями с разделением во времени при использовании площадки для производства разной продукции.
Фильтры для стерилизации жидкостей, как правило, предназначены для однократного использования при производстве одной серии или для одной производственной компании (когда один и тот же фильтр применяют для нескольких серий на основе одной компании); однако, некоторые из них могут предназначаться для многократного использования (когда фильтр очищают и повторно стерилизуют между сериями продукции). Фильтры не следует использовать повторно для жидкостей, отличных по химическому составу, за исключением случаев, когда проведена валидация в отношении выделения вымываемых веществ и абсорбции после надлежащей очистки и стерилизации фильтра.
Если стерилизующий фильтр используют в заключительном этапе стерилизующей фильтрации для получения стерильного жидкого фильтрата, как правило, не рекомендуется его повторное использование. Повторное использование стерилизующего фильтра может считаться целесообразным в тех случаях, когда фильтруемая жидкость имеет низкий уровень бионагрузки и когда:
a) фильтруют разные серии одного и того же жидкого продукта, и
b) общий объем фильтрата не влияет на производительность фильтра.
Примечание 1 - Регуляторные органы не запрещают повторное использование стерилизующих фильтров для жидкостей, однако обычно не поддерживают такую практику при асептическом производстве вследствие критического характера процесса стерилизующей фильтрации. Дополнительные сложности, связанные с повторным использованием фильтров, могут привести к несоблюдению нормативных требований и дополнительным регуляторным действиям в отношении производителя продукции.
Примечание 2 - Стерилизующий фильтр может быть использован для обеспечения требуемого уровня содержания механических частиц или бионагрузки перед этапом стерилизующей фильтрации. При этом фильтрат, полученный с использованием такого фильтра, не считают стерильным.
Следует принимать во внимание, что если на упаковке фильтра указано, например, что стерилизующий фильтр выдерживает 25 циклов стерилизации паром при 121 °C в течение 20 мин, не следует делать однозначный вывод, что фильтр может быть повторно стерилизован и использован для стерилизующей фильтрации жидкости до 25 раз. Данное утверждение может содержать только информацию об устойчивости фильтра с точки зрения его запаса прочности, связанной с количеством циклов стерилизации, которые фильтр может безопасно выдержать, прежде чем возникнет проблема с разрушением полимера. Эта информация об обработке фильтра может оказаться важной для производителя асептической продукции в отношении проведения повторной стерилизации фильтра, однако ее не следует экстраполировать на возможность многократной повторной стерилизации и возможность повторного использования фильтра.
Рекомендации приведены для 8.2.3.2 b).
При проведении валидационных и квалификационных работ по подтверждению того, что повторное использование стерилизующего фильтра в процессе фильтрации определенной жидкости не ухудшает его производительность или качество фильтрата, следует учитывать следующее, но не ограничиваться этим:
a) любое воздействие на фильтр, связанное с процессами, которые выполняет пользователь фильтра (например, многократная фильтрация, промывка, очистка, ополаскивание, сушка и стерилизация) при его повторном использовании;
b) условия хранения повторно используемых фильтров, в том числе после непосредственного использования и после стерилизации перед повторным использованием;
c) вымывание контаминантов или остатков моющего средства из использованных фильтров. Следует учитывать возможность контаминации бактериальным эндотоксином в результате лизиса бактериальных клеток при переносе бионагрузки из одной серии в другую.
Валидационные исследования эффективности задерживания микроорганизмов следует проводить на фильтрах, предназначенных для повторного использования и подвергавшихся воздействию реальных производственных условий (включая производственные условия наихудшего случая). Данные исследования необходимы, поскольку испытания на целостность фильтра после использования при отсутствии исследований эффективности задерживания бактерий недостаточно для определения того, влияет ли многократное повторное использование на эффективность стерилизующего фильтра.
А.8.3 Процесс фильтрации
Рекомендации отсутствуют.
А.8.4 Испытание фильтра на целостность
А.8.4.1 Рекомендации отсутствуют.
А.8.4.2 Испытание на целостность фильтра перед фильтрацией возможно не проводить, если:
a) фильтрат однозначно отбраковывается в случае отрицательного результата испытания на целостность фильтра после использования, или
b) результаты оценки риска процесса стерилизации допускают повторную фильтрацию.
На рисунках А.11 и А.12 приведены примеры схем, используемых при испытаниях на целостность фильтра после стерилизации перед использованием, предназначенные для обеспечения стерильности системы.
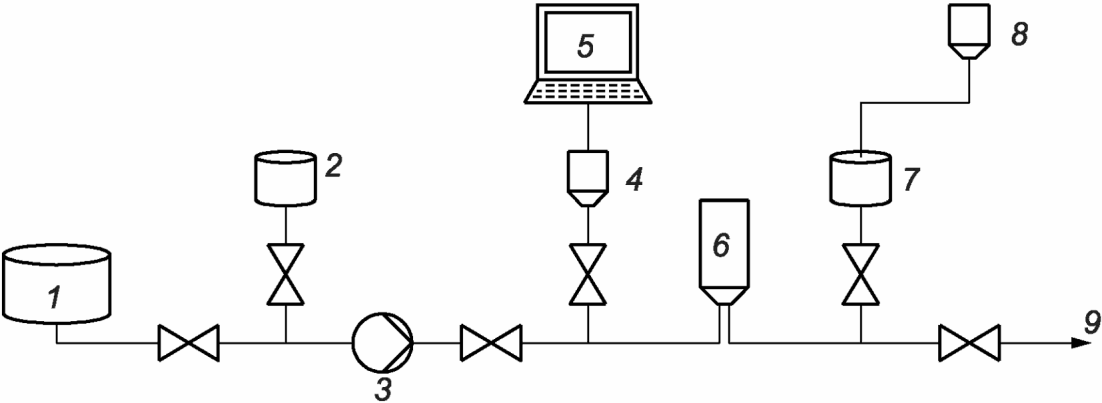
1 - продукт; 2 - смачивающая/промывающая жидкость;
3 - насос; 4 - фильтр для газов с размером пор 0,2 мкм;
5 - прибор для испытания на целостность фильтра; 6 - фильтр
с размером пор 0,2 мкм для стерилизации продукта;
7 - биоконтейнер для смачивающей/промывающей жидкости;
8 - фильтр для газов с размером пор 0,2 мкм;
9 - стерильный продукт
испытания на целостность одного фильтра с размером пор
0,2 мкм перед использованием в предварительно
стерилизованной системе для однократного использования
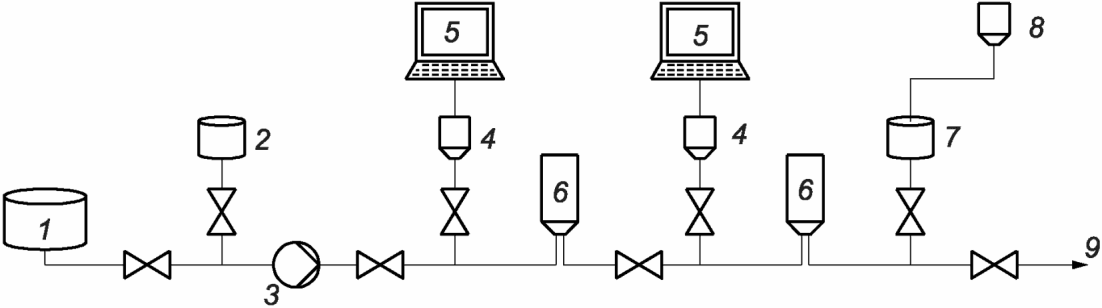
1 - продукт; 2 - смачивающая/промывающая жидкость;
3 - насос; 4 - фильтр для газов с размером пор 0,2 мкм;
5 - прибор для испытания на целостность фильтра; 6 - фильтр
с размером пор 0,2 мкм для стерилизации продукта;
7 - биоконтейнер для смачивающей/промывающей жидкости;
8 - фильтр для газов с размером пор 0,2 мкм;
9 - стерильный продукт
испытания на целостность перед использованием двух отдельных
фильтров с размером пор 0,2 мкм в предварительно
стерилизованной системе для однократного использования
А.8.4.3 Рекомендации отсутствуют.
А.8.4.4 Рекомендации отсутствуют.
А.8.4.5 Рекомендации отсутствуют.
А.8.4.6 Рекомендации отсутствуют.
А.9 Валидация
А.9.1 Общие положения
Рекомендации отсутствуют.
А.9.2 Валидация эффективности задерживания микроорганизмов, специфичных для фильтруемой среды, стерилизующими фильтрами для жидкостей
А.9.2.1 Общие положения
А.9.2.1.1 Рекомендации отсутствуют.
А.9.2.1.2 В предыдущих изданиях настоящего стандарта допускалось отклонение результатов испытания на физическую целостность одного из испытуемых фильтров не более чем на 10% от диапазонов значений, установленных производителем фильтра. В связи с усовершенствованием производства фильтров в настоящее время отклонение результатов испытания на целостность фильтра от среднего значения по спецификации более чем на 40% практически невозможно.
А.9.2.1.3 Рекомендации отсутствуют.
А.9.2.1.4 Рекомендации отсутствуют.
А.9.2.1.5 Рекомендации отсутствуют.
А.9.2.2 Типовые микроорганизмы (тест-штаммы микроорганизмов)
А.9.2.2.1 Рекомендации отсутствуют.
А.9.2.2.2 Рекомендации отсутствуют.
А.9.2.2.3 При культивировании B. diminuta условия культивирования могут приводить к уменьшению размера клеток и, соответственно, повышению способности микроорганизмов проходить через фильтр [8].
Для выявления микроорганизмов малого размера в производственной среде и исходных материалах обычно проводят анализ штаммов, выделенных в ходе мониторинга производственной среды, контроля исходных материалов и продукта перед его фильтрацией. При необходимости размеры микроорганизмов штаммов, выделенных из фильтруемой среды, сравнивают с B. diminuta.
Использование воды из систем водоподготовки, не снабженных системами дезинфекции, может способствовать присутствию микроорганизмов малого размера.
А.9.2.2.4 Рекомендации отсутствуют.
А.9.2.2.5 Рекомендации отсутствуют.
А.9.2.2.6 Культивирование A. laidlawii в соево-казеиновом бульоне <*> (TSB <**>) может привести к заметному увеличению процента бактериальных клеток меньшего размера, однако эти клетки могут обладать меньшей проникающей способностью по сравнению с клетками, выращенными в других условиях. Питательная среда с добавкой 10% лошадиной сыворотки может способствовать получению клеток с большей проникающей способностью (которые не обязательно будут иметь малый размер) [9].
--------------------------------
А.9.2.2.7 Рекомендации отсутствуют.
А.9.2.2.8 Рекомендации отсутствуют.
А.9.2.2.9 Рекомендации отсутствуют.
А.9.2.2.10 Рекомендации отсутствуют.
А.9.3 Валидация методики испытания на целостность стерилизующих фильтров для жидкостей
Рекомендации отсутствуют.
А.9.4 Валидация процесса взаимодействия фильтра с фильтруемой средой
Рекомендации отсутствуют.
А.9.5 Валидация процесса стерилизации системы фильтрации
А.9.5.1 При рассмотрении процесса стерилизации стерилизующего фильтра следует учитывать возможность того, что данный процесс может привести к повреждению фильтра или системы фильтрации. Например, стерилизация паром на месте может быть использована для устранения или уменьшения количества асептических соединений; однако, при этом может возникнуть риск повреждения мембраны фильтра вследствие прохождения изменяющегося потока пара через фильтр при повышенной температуре. Снижение риска физического повреждения мембраны фильтра может быть достигнуто в результате применения статических процессов, таких как радиационная стерилизация или стерилизация влажным теплом в паровом стерилизаторе; однако использование перечисленных подходов может потребовать использования большего количества асептических соединений.
А.9.5.2 Рекомендации отсутствуют.
А.9.6 Валидация эффективности задерживания микроорганизмов, специфичных для фильтруемой среды, стерилизующими фильтрами для газов
А.9.6.1 Общие положения
Фильтры для газов, используемые в качестве "дыхательных", применяют в асептическом производстве, в том числе для реакторов, автоклавов и лиофилизаторов, азотной подушки, защищающей чувствительную к кислороду продукцию, и оборудования продувки-фасовки-герметизации.
А.9.6.2 Задерживание аэрозоля микроорганизмов
Для оценки задерживающей способности фильтров для газов могут быть проведены испытания с использованием типовых микроорганизмов в жидкой или аэрозольной взвеси. Использование типовых микроорганизмов в жидкой взвеси является наихудшим случаем, поскольку эффективность задерживания фильтров для газов при фильтрации жидкости намного ниже, чем при фильтрации газов. С другой стороны, эффективность задерживания микроорганизмов в газе будет более высокой, поэтому использование типовых микроорганизмов в аэрозольной взвеси является менее строгим по сравнению с жидкими взвесями, несмотря на то, что имитирует предполагаемые условия использования.
А.9.6.3 Валидация методики испытания на физическую целостность фильтра
Рекомендации отсутствуют.
А.9.6.4 Совместимость и срок эксплуатации
Рекомендации отсутствуют.
А.9.6.5 Валидация процесса стерилизации системы фильтрации для газов
Рекомендации отсутствуют.
А.10 Текущий мониторинг и контроль
Рекомендации отсутствуют.
А.11 Выпуск в обращение продукции, прошедшей стерилизующую фильтрацию
Рекомендации отсутствуют.
А.12 Поддержание эффективности процесса
А.12.1 Общие положения
Рекомендации отсутствуют.
А.12.2 Повторная калибровка
Рекомендации отсутствуют.
А.12.3 Техническое обслуживание оборудования
Рекомендации отсутствуют.
А.12.4 Повторная квалификация (валидация)
Рекомендации отсутствуют.
А.12.5 Оценка изменений
Если изменяют фильтруемую среду (например, изменение исходных материалов, химического состава, концентрации, вязкости, изменение условий процесса стерилизации), то необходимо оценить, могут ли эти изменения оказать негативное влияние на эффективность стерилизующего фильтра или нарушить валидность используемого процесса стерилизующей фильтрации.
(справочное)
НАЦИОНАЛЬНЫМ И МЕЖГОСУДАРСТВЕННЫМ СТАНДАРТАМ
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального, межгосударственного стандарта |
ISO 11135 | IDT | ГОСТ ISO 11135-2017 "Стерилизация медицинской продукции. Этиленоксид. Требования к разработке, валидации и текущему управлению процессом стерилизации медицинских изделий" |
ISO 11137-1 | IDT | ГОСТ ISO 11137-1-2011 "Стерилизация медицинской продукции. Радиационная стерилизация. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" |
ISO 11139 | - | |
ISO 13408-1:2008 | - | |
ISO 13408-5 | IDT | ГОСТ Р ИСО 13408-5-2011 "Асептическое производство медицинской продукции. Часть 5. Стерилизация на месте" |
ISO 13485 | IDT | ГОСТ ISO 13485-2017 "Изделия медицинские. Системы менеджмента качества. Требования для целей регулирования" |
ISO 17665-1 | IDT | ГОСТ Р ИСО 17665-1-2016 "Стерилизация медицинской продукции. Влажное тепло. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" |
<*> Соответствующий национальный/межгосударственный стандарт отсутствует. До его принятия рекомендуется использовать перевод на русский язык данного международного стандарта. Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: - IDT - идентичные стандарты. | ||
[1] | ISO 10993-12:2012 | Biological evaluation of medical devices - Part 12: Sample preparation and reference materials |
[2] | ISO 13408-3 | Aseptic processing of health care products - Part 3: Lyophilization |
[3] | ISO 13408-4 | Aseptic processing of health care products - Part 4: Clean-in-place technologies |
[4] | ISO 13408-6 | Aseptic processing of health care products - Part 6: Isolator systems |
[5] | ISO 13408-7 | Aseptic processing of health care products - Part 7: Alternative processes for medical devices and combination products |
[6] | ISO 11737-1 | Sterilization of medical devices - Microbiological methods - Part 1: Determination of a population of microorganisms on products |
[7] | ISO/IEC 90003 | Software engineering - Guidelines for the application of ISO 9001:2008 to computer software |
Lee S-H, Lee S-S, Kim C-W Changes in the Cell Size of Brevundimonas diminuta Using Different Growth Agitation Rates. PDA J. Pharm. Sci. Technol. 2002, 56 pp. 99 - 108 | ||
Parenteral Drug Association. PDA Technical Report No. 75 (TR 75) Consensus Method for Rating | ||
УДК 637.132.4:715.478:658.513:006.354 | ОКС 11.080.01 |
Ключевые слова: асептическое производство, медицинская продукция, стерилизующая фильтрация, стерилизующий фильтр, валидация | |