СПРАВКА
Источник публикации
М.: Стройиздат, 1973
Примечание к документу
Название документа
"Руководство по химическому и технологическому анализу воды"
"Руководство по химическому и технологическому анализу воды"
Содержание
ПО ХИМИЧЕСКОМУ И ТЕХНОЛОГИЧЕСКОМУ АНАЛИЗУ ВОДЫ
Руководство содержит методы химического и технологического анализа воды для персонала лабораторий, работающих в области водоснабжения промышленных предприятий и населенных мест.
Изложенные в книге методы химического и технологического анализа должны способствовать рациональному использованию и охране от загрязнения водных ресурсов, а также повышению эффективности контроля качества вод для питьевого и промышленного водоснабжения.
Руководство предназначено для работников лабораторий, занимающихся анализом воды.
Нормативные документы по методам химических и технологических анализов вод, используемых для хозяйственно-питьевого и промышленного водоснабжения, были изданы в виде отдельных Государственных общесоюзных стандартов в 1945 - 1949 гг. С тех пор появились новые методы анализа, разработаны новые реактивы, позволяющие повысить точность анализов и ускорить их проведение, возникла необходимость в дополнительном определении некоторых ингредиентов природных вод, для которых не имелось ГОСТов. Все это привело к необходимости разработки нового руководства по методам химических и технологических анализов воды для персонала лабораторий, работающих в области водоснабжения промышленных предприятий и населенных мест.
Из практики проектирования сооружений по подготовке воды для промышленного и хозяйственно-питьевого водоснабжения вытекает необходимость технологических анализов для обоснования выбора методов подготовки и определения основных расчетных параметров водоподготовительных сооружений. Данные технологических анализов нужны также для предварительного определения условий эксплуатации водоподготовительных установок, с последующим их уточнением в условиях промышленного использования сооружений.
Настоящее Руководство состоит из двух разделов. Первый раздел составлен ВНИИ ВОДГЕО в основном по методикам, одобренным Совещанием руководителей водохозяйственных органов стран - членов Совета экономической взаимопомощи ("Унифицированные методы исследования качества вод" изд. СЭВ, Москва, 1965 г.).
Включенные во второй раздел Руководства методики технологических анализов разработаны: Институтом коммунального водоснабжения и очистки воды Академии коммунального хозяйства им. К.Д. Памфилова ("Определение доз коагулянтов, необходимых для осветления и обесцвечивания воды", "Определение необходимых доз флокулянтов", "Определение основных показателей работы контактных осветителей", "Определение фильтровальных характеристик воды и расчет фильтрующих загрузок", "Выбор метода обезжелезивания воды"), ВНИИ ВОДГЕО ("Определение осаждаемости взвесей", "Определение доз хлора", "Определение стабильности воды").
Замечания по Руководству следует направлять по адресу: Москва, Г-48, Комсомольский проспект, д. 42, ВНИИ ВОДГЕО (Индекс 119826).
Общие положения
1.1. Правильный отбор пробы воды является важной частью ее анализа, необходимым условием надежности получаемых результатов. Ошибки, возникающие вследствие неправильного отбора пробы, в дальнейшем исправить нельзя.
При отборе пробы следует учитывать многие обстоятельства. Выбор способа и само взятие пробы должны производить опытные квалифицированные работники.
Условия, которые следует соблюдать при отборе проб, настолько разнообразны, что нельзя дать подробных рекомендаций для всех случаев. Поэтому здесь приводятся лишь общие указания и даны наиболее важные общие принципы. Главные принципы, которые требуется соблюдать при отборе проб воды, состоят в следующем:
а) проба воды, взятая для анализа, должна отражать условия и место ее взятия (см. стр. 5 и 8);
б) отбор пробы, ее хранение, транспортировка и обращение с ней должны производиться так, чтобы не произошли изменения в содержании определяемых компонентов или в свойствах воды (см. стр. 11);
в) объем пробы должен быть достаточным и должен соответствовать применяемой методике анализа (см. стр. 6).
Техника отбора пробы
Выбор места для отбора
1.2. Место для отбора пробы выбирается соответственно цели анализа и на основании обследования местности, причем учитываются все обстоятельства, которые могли бы оказать влияние на состав взятой пробы воды.
Виды отбора проб
1.3. Соответственно цели анализа применяются разовый или серийный отбор проб.
При разовом отборе пробу берут один раз в определенном месте и рассматривают результат одного анализа. Этот способ применяется в редких случаях, когда результатов одного анализа достаточно для суждения о качестве исследуемой воды (например, при постоянстве качества воды, как это наблюдается в глубинных грунтовых водах).
Ввиду того, что качество воды в большинстве случаев изменяется как в разных местах данного объекта, так и в различные периоды времени, однократного взятия пробы воды обычно недостаточно. В таких случаях применяют серийный отбор проб, при котором каждая проба берется в определенной связи с остальными пробами. При анализе серии взятых проб определяется изменение содержания наблюдаемых компонентов с учетом места, времени или обоих этих факторов. Анализом серии проб получают соответствующее количество результатов, которые обрабатывают и расценивают обычно с использованием методов математической статистики.
Типичным примером серийного отбора проб является зональный отбор. Пробы отбирают с различных глубин по выбранному створу водохранилища, озера, пруда. Следующий, весьма распространенный тип серийного отбора проб - отбор через определенные промежутки времени. Такой отбор позволяет следить за изменением качества воды во времени или же в зависимости от ее расхода.
Обычно отбирают ряд проб для определения сезонных или дневных изменений качества воды, т.е. в интервалах месяцев, суток или часов. Часто производят суточный отбор проб, при котором ряд проб отбирают в течение суток через каждые 1 - 3 ч.
Особый тип серийного отбора представляют так называемые "согласованные пробы", которые отбирают в различных местах по течению реки с учетом времени прохождения воды от одного пункта до другого.
Виды проб
1.4. Различают две основные пробы: простую и смешанную.
Простую пробу получают путем отбора всего требуемого количества за один раз. Анализ простой пробы дает сведения о составе воды в данный момент в одном месте.
Смешанную пробу получают, сливая одинаковые по объему простые пробы, взятые из одного и того же места несколько раз подряд через определенные промежутки времени или отобранные одновременно из различных мест обследуемого объекта. Эта проба должна характеризовать средний состав воды исследуемого объекта, или средний состав за определенный период времени (за час, за смену, за день), или, наконец, средний состав с учетом как места, так и времени.
Среднюю пробу приготовляют обычно смешением равных частей проб, отобранных через равные промежутки времени. Однако этот простой способ пригоден только в том случае, если все точки исследуемого объекта равноценны или если в месте отбора проб имеется постоянный расход воды. Если же это не так, то приготовляют среднюю пропорциональную пробу соединением различных объемов проб, взятых в равных интервалах времени, или равных объемов проб в различных интервалах времени таким образом, чтобы их объем или число соответствовали местным колебаниям или изменениям расхода.
Средняя проба тем точнее, чем меньше интервалы между отдельно взятыми составляющими ее пробами. Наилучший результат можно получить, автоматизируя непрерывный отбор проб.
Смешанную пробу не рекомендуется отбирать за период больше одних суток. Она должна быть предохранена от качественных изменений, вызываемых длительным хранением (см. стр. 11). При консервировании проб соответствующий реагент прибавляют в сосуд для взятия пробы заранее с той целью, чтобы все составляющие пробы сразу же после взятия смешивались с реагентом. Смешанную пробу нельзя применять для определения компонентов и характеристик воды, легко изменяющихся (растворенные газы, pH). Если же их определение необходимо производить, то оно делается в каждой составляющей пробе отдельно. Смешанную пробу нельзя составлять и в том случае, если характер воды изменяется во времени в такой степени, что отдельные составляющие пробы вступают во взаимодействие или изменяют состав воды вследствие изменения их физического состояния.
Количество пробы, необходимое для анализа
1.5. Количество пробы, которое необходимо отобрать, зависит от числа определяемых компонентов. Для неполного анализа, при котором определяются только несколько компонентов или свойств воды: гигиеническая оценка, некоторые контрольные определения и т.д. (см. стр. 11) - достаточно отобрать 1 л воды. Для более подробного анализа следует брать 2 л. Для полного анализа или для определения компонентов, которых очень мало в воде, требуется еще больший объем пробы. (В описании хода анализа при определении различных компонентов даны указания о требуемом объеме воды.)
Сосуды для отбора и хранения пробы
1.6. Чаще всего используются бутыли из прозрачного, бесцветного, химически стойкого стекла или бутыли и другие сосуды из полиэтилена с притертой стеклянной пробкой или со специальными пробками, имеющими пружинные крепления, с резиновым уплотнением.
Для основной пробы обычно применяется бутыль емкостью 2 л. Большие пробы отбирают в бутыли емкостью 20 - 25 л, снабженные резиновыми пробками и защищенные предохранительными корзинами. Пробы, содержащие крупные примеси, в особенности смешанные пробы, отбирают в широкогорлые банки или канистры. Дополнительные пробы для проведения некоторых определений, требующих специальной обработки (см. стр. 13 - 20), отбирают в меньшие бутыли с притертыми или резиновыми пробками или же в кислородные склянки.
Используемую для проб посуду следует предварительно тщательно вымыть. Для мытья стеклянных и полиэтиленовых бутылей в настоящее время применяют концентрированную соляную кислоту (техническую), для обезжиривания используют синтетические моющие вещества. Сильно загрязненные стеклянные бутыли моют и обезжиривают хромовой смесью (к 35 мл насыщенного водного раствора бихромата калия осторожно приливают, перемешивая, 1 л концентрированной серной кислоты; реактивы применяют технические). Полное обезжиривание достигается путем пропаривания перевернутой посуды водяным паром. Остатки использованного для мытья реактива должны быть полностью удалены тщательной промывкой бутылей обыкновенной водой. Наконец, вымытую посуду ополаскивают дистиллированной водой, дают воде стечь и, если надо, высушивают.
Прежде чем взять пробу, посуду следует ополоснуть несколько раз отбираемой водой. Бутыли, наполненные пробой, нужно надписать или пронумеровать.
Приборы и приспособления для отбора проб
1.7. В большинстве случаев можно взять пробу прямо в бутыль. Если доступ к воде затруднен, проба отбирается батометром.
Для определения некоторых веществ весьма важно, чтобы проба воды при взятии ее была защищена от соприкосновения с атмосферным воздухом. При отборе пробы непосредственно бутылью следует избегать перемешивания воды с воздухом, выходящим из погружаемой бутыли. Этого можно достичь применением насадки. Она представляет собой резиновую пробку, в которую вставлены две стеклянные трубки, одна из них оканчивается у дна бутыли, другая - у пробки. Бутыль, снабженная такой насадкой, наполняется водой равномерно без взмучивания.
Если отбор проб производится при помощи пробоотборного сосуда, воду не переливают прямо в бутыль, а применяют для этого сифонную трубку (резиновый шланг), которую опускают до дна бутыли. После наполнения последней сифонную трубку оставляют еще на некоторое время, чтобы вода перетекала через края, и только после этого закрывают пробкой так, чтобы в бутыли не оставались пузырьки воздуха. Если пробы отбирают при помощи глубинных приборов, то воду выпускают из них через резиновый шланг, надетый на выпускной кран и опущенный на дно бутыли. И в этом случае вода должна перетекать некоторое время через края бутыли. Подобным же образом используется резиновый шланг и при отборе проб из водопроводных кранов.
Если требуется полностью предохранить пробу от соприкосновения с атмосферным воздухом, ее следует отбирать таким образом, чтобы она не соприкасалась даже с воздухом, находящимся в сосуде для пробы. При взятии проб с глубины при помощи пробоотборных приборов это условие соблюдается. Однако при взятии проб быстротекущей воды или воды из мелких водоемов нельзя использовать глубинные пробоотборные приборы. Для этой цели пользуются горизонтальным пробоотборным прибором, сконструированным подобно глубинному, но с тем различием, что главная ось прибора проходит горизонтально, или же используют различные комбинации сообщающихся сосудов. Принцип устройства последних состоит в том, что вода, наполнившая первую бутыль, переливается через соединительную трубку в другую бутыль большего объема, погруженную вместе с первой под воду. Первая бутыль уже не содержит воздуха. Пока наполняется вторая, большая бутыль, содержимое первой сменяется несколько раз и последующие порции с воздухом не соприкасаются. Первая бутыль, наполненная таким способом без соприкосновения с воздухом, может служить непосредственно для хранения пробы.
Если пробу воды нельзя отбирать сосудом вследствие, например, слишком мелкого водоема или недоступности места отбора, пробу откачивают насосом с ручным приводом или мотором.
Запись о взятии проб
1.8. Цель записи отбора каждой пробы состоит в точном учете условий отбора. В записи следует указать вид и происхождение воды, точное место отбора (описание, план места), день и час отбора и номера отдельных бутылей с пробами.
Запись по мере надобности дополняют кратким результатом исследования, произведенного на месте; описанием примененного способа взятия, способа консервирования и всех важных обстоятельств, установленных при отборе проб (метеорологические условия во время отбора и при необходимости также в предшествующие дни, изменения, происшедшие в окружающей местности, состояние производства на заводе и пр.). При повторном отборе проб с одного и того же места кратко отмечаются только условия и место отбора и не повторяется детальное описание, план и фотография места отбора.
Основные указания для отбора проб воды
из различных источников
Взятие проб из рек и ручьев
1.9. Усредненную пробу протекающей воды берут в местах наиболее сильного течения, т.е. лучше всего в фарватере течения. Если цель исследования не указана, рекомендуется избегать отбора проб стоячей воды перед плотинами, в подпорах, в изгибах, глухих рукавах и т.д. Фарватера можно достигнуть на лодке, с моста или же с обрывистого берега на излучине, где фарватер прижат к берегу. Пробу берут под поверхностью воды, лучше всего в верхней трети общей глубины (приблизительно 20 - 30 см под поверхностью). Пробы отбираются единовременно или серийно, простые или смешанные.
Часто требуется отразить в пробе результаты смешения двух потоков или речной воды со сточными водами. В этом случае пробу следует отбирать в месте полного смешения вод обоих потоков. Определить место полного смешения довольно трудно. Надо иметь в виду, что смешение вод различной плотности и различной температуры происходит очень медленно. Оно зависит от скорости течения, рельефа русла реки, количества изгибов, порогов. Обычно полное смешение происходит на расстоянии сотен метров или нескольких километров от места слияния потоков. В свою очередь, место взятия пробы не должно быть слишком далеко от места слияния потоков, так как следует учитывать и изменения качества воды, происходящие во времени. Также следует учитывать то, что между мостом слияния потоков и пунктом полного смешения могут оказаться плотины, притоки и последующие сбросы сточных вод. Надо также иметь легкий доступ к месту взятия пробы в течение всего года. Не рекомендуется менять выбранное место, если оно удовлетворяет необходимым условиям.
Для некоторых специальных исследований иногда требуется установить различие в качестве проб, взятых из разных мест поперечного сечения потока. В этом случае выбирают несколько вертикалей, по которым отбирают пробы с различных глубин данного створа. Такой отбор обычно сопровождается измерением скорости течения в отдельных пунктах взятия проб.
На подпертых участках, а также непосредственно под плотиной пробы отбирать не следует, так как такие пробы не точно отражают содержание в воде растворенных газов и некоторых других компонентов.
Каждый отбор пробы воды из потока должен быть дополнен измерением расхода по соответствующему профилю в момент отбора пробы. Поэтому целесообразно выбирать места для отбора проб, расположенные вблизи гидрометрического поста или водомерной рейки.
Отбор проб из водохранилища, озер и прудов
1.10. Стоячие воды обычно неоднородны и различаются по качеству воды в различных местах и на различных глубинах. Поэтому следует отбирать пробы из различных мест и разных глубин.
Не рекомендуется отбирать так называемую среднюю пробу из водохранилища, т.е. получать пробу смешением пропорциональных порций воды, взятых из различных мест всего водохранилища. Вследствие существенных различий качества воды в различных местах компоненты проб могут вступить во взаимодействие и совершенно исказить истинное представление о качестве воды.
При отборе проб стоячей воды следует избегать мест с густыми порослями водяных растений. Качество воды, вытекающей из водохранилища, лучше всего определять в пробе, взятой на соответствующей глубине непосредственно перед падением воды, а не из самого каскада.
Отбор проб из родников, колодцев, скважин и дренажей
1.11. Если вблизи источника построен искусственный водоприемник, пробу берут из него под поверхностью воды непосредственно в бутыль пробоотборным прибором. Если источник снабжен сливной трубкой или желобком, пробу отбирают в сосуд прямо со сливной трубки. Если требуется предотвратить потерю растворенных газов, конец трубки или желобка погружают в большую воронку, которую наполняют водой до краев, выжидают некоторое время, в течение которого вода переливается через край, и затем длинную трубку воронки опускают до дна бутылки для пробы. После наполнения бутыли дают воде некоторое время перетекать через края бутыли.
Если у источника нет водоприемника и нет искусственного слива, то пробу текущей воды берут непосредственно в сосуд или с помощью насоса. Иногда родник надо предварительно очистить. Делают это за день до взятия пробы. Дно в непосредственной близости ключа углубляют таким образом, чтобы в углубление можно было свободно погрузить бутыль для пробы или другую посуду. После дождя отбор проб целесообразно производить из скважин одновременно с опытной откачкой, чтобы можно было установить постоянное качество воды и выявить, не загрязняется ли она поверхностными водами.
При отборе проб из колодца сначала откачивают из него воду. Если последний в течение долгого времени эксплуатировался мало, откачивание воды продолжают до тех пор, пока выкачиваемая вода не будет иметь постоянную температуру (примерно 20 мин). При этом следят за тем, чтобы выкачиваемая вода стекала подальше от колодца и не могла проникнуть обратно в него. Только после этого приступают к наполнению бутыли для пробы или пробоотборного сосуда. Чтобы предотвратить потерю растворенных газов, пользуются воронкой, в которую погружают конец выходной трубки насоса.
Если колодец в течение долгого времени не эксплуатировался, выкачивают из него воду и оставляют до тех пор, пока он снова не наполнится. Если колодец не оборудован стационарным насосом, воду выкачивают передвижным насосом. После этого можно производить отбор пробы.
Отбор проб из колодцев лучше всего производить в летнее время при сухой погоде, когда расход воды и ее обмен максимальны.
При отборе проб из колодца следует учитывать все необычные обстоятельства, например недавнее окончание постройки или ремонта колодца, его дезинфекцию.
Пробы из скважин отбираются глубинным пробоотборником с узким сечением или насосом. Пробы из скважин, в которых долго стояла вода или верхнее отверстие которых не было достаточно закрыто, не являются надежными для анализа.
Пробы воды из дренажей отбирают непосредственно из стока дренажных труб. Для дренажных канав, в которых нет дренажных труб и где вода стекает по дну, применяют чистые, лучше керамические трубы длиной около 1 м. Трубу укладывают в канаву так, чтобы через нее протекала часть воды. Пробоотборный сосуд подставляют к концу трубы и наполняют его.
Отбор проб из водопроводных станций, сети
и водоразборных кранов
1.12. На водопроводных станциях пробы берут из выходной трубки насосов или из сборных желобов. При отборе из резервуара пробу берут под поверхностью воды. Из всасывающего или сифонного трубопровода воду откачивают в сосуд вакуум-насосом. В тех местах водопроводной сети, в которых пробы отбирают регулярно, рекомендуется вмонтировать постоянный кран для взятия проб. На всасывающем трубопроводе следует смонтировать короткий патрубок с запорным вентилем, краном, вторым краном и снова запорным вентилем. Перед взятием пробы оба вентиля закрывают и таким образом изолируют часть воды, находящуюся в патрубке, от остального трубопровода. Под нижний кран подставляют сосуд для пробы, и оба крана открывают. Через верхний кран входит воздух, а вода вытекает из нижнего.
Из водопроводных кранов пробы берут следующим образом. На кран надевают резиновый шланг, второй конец которого вводят в бутыль для пробы, опуская его до дна. Открывают медленно кран, пока вода не потечет непрерывной струей толщиной около 0,5 см. После наполнения сосуда водой его оставляют еще некоторое время под краном, чтобы вода перетекала через края сосуда до тех пор, пока температура воды не будет постоянной. Если требуется определить содержание токсичных веществ в водопроводной воде (медь, свинец), то пробу берут сразу же после открытия крана. Тогда в пробу поступает та часть воды, которая долго оставалась в трубопроводе (например, в течение ночи).
Консервирование проб
1.13. Консервирование проб воды преследует цель сохранения компонентов, определяемых в воде, и ее свойств в том состоянии, в каком они находились в ней в момент взятия пробы. Консервирование необходимо особенно в тех случаях, когда определяемый компонент подвергается изменениям и когда соответствующее определение нельзя произвести сразу же на месте отбора пробы и в тот же день в лаборатории. Соответственно степени изменяемости воды и относительной чистоты пробы (и если проба не была консервирована) определение производится:
б) как можно раньше, не позже чем через 2 ч после взятия пробы;
в) в тот же день; приступают к анализу в день отбора пробы, не позже чем через 12 ч после ее отбора;
г) через более продолжительное время, чем было указано в пунктах "а" и "в".
За время, которое прошло между отбором пробы и определением, исследуемые вещества могут изменяться в различной степени. Очень быстро изменяются температура воды и ее pH. Газы, содержащиеся в воде, например кислород, двуокись углерода, сероводород или хлор, могут улетучиться из пробы или появиться в ней (кислород, двуокись углерода). Эти и подобные им вещества надо определять на месте отбора пробы или фиксировать. Изменение равновесия системы (величины pH, содержания карбонатов, свободной двуокиси углерода) может вызвать изменение других компонентов, содержащихся в пробе. Некоторые из них могут выделяться в виде осадка или, наоборот, из нерастворимой формы перейти в раствор. Это относится особенно к солям железа, марганца, кальция.
В неконсервированной пробе обычно протекают различные биохимические процессы, вызванные деятельностью микроорганизмов или планктона. Эта метаболическая деятельность протекает в отобранной пробе иначе, чем в первоначальной среде, и ведет к окислению или восстановлению некоторых компонентов пробы: нитраты восстанавливаются до нитритов или до ионов аммония, сульфаты - до сульфидов, или, наоборот, происходит окисление сульфидов, сульфитов, двухвалентного железа, цианидов. Влияние различных факторов на изменение компонентов, содержащихся в воде, может происходить непосредственно или косвенным путем. Органолептические свойства воды, например запах и привкус, также могут изменяться. Равным образом изменяются цвет, мутность и прозрачность воды. Некоторые компоненты могут адсорбироваться на стенках бутыли (железо, медь, кадмий, алюминий, марганец, хром, цинк, фосфаты) или выщелачиваться из стекла или пластмассы бутыли (бор, кремний, натрий, калий; различные ионы, адсорбированные полиэтиленом при предшествующем использовании бутыли).
Нельзя указать общее правило или привести единые нормы, в какой срок должно быть осуществлено определение того или иного компонента или каким способом следует произвести консервирование пробы. Правильность результата химического анализа зависит от опыта аналитика и в значительной мере от оценки полученных им данных. Промежуток времени между взятием пробы и ее анализом зависит от характера пробы, рода производимого анализа и условий хранения пробы. В общем можно сказать, что чем больше вероятность изменения компонента, подлежащего определению, чем больше вода загрязнена (при отсутствии в ней токсических веществ), тем раньше следует произвести анализ. Применение консервирующих средств не предохраняет полностью определяемое вещество от изменения, вследствие чего и консервированные пробы следует анализировать в соответствующее время, обычно на следующий день, но не позднее чем на третий день после отбора пробы.
Дата отбора пробы и дата начала анализа должны быть указаны в протоколе анализа. Точно так же следует указать и способ консервирования. При вычислении концентрации определяемого компонента следует учитывать возможные изменения объема пробы, вызванные прибавлением консервирующего вещества.
Универсального консервирующего вещества не существует. Для полного анализа воды обычно требуется отобрать пробу в несколько бутылей, которые консервируются различными консервирующими веществами. Для определения содержания некоторых компонентов, например сульфидов, сульфитов, агрессивной двуокиси углерода, следует брать пробы в отдельные бутыли для каждого из этих определений.
В некоторых случаях перед определением консервированную пробу следует нейтрализовать. Для отделения растворенных веществ от нерастворенных пробу фильтруют, лучше всего на месте ее отбора.
В табл. 1 приведен список отдельных компонентов и указаны возможности условий консервирования, способы отбора пробы и допустимые интервалы между отбором пробы и началом анализа. Соблюдение этих условий наиболее надежно задержит изменение определяемого компонента.
Таблица 1
Компонент | Указания о консервировании пробы |
Агрессивная CO2 | Нельзя консервировать, см. ниже "Кислотность". Хранение при температуре 3 - 4 °C. Пробу анализируют сразу после отбора. Проба с мрамором: отбор пробы в бутыль, которая содержит CaCO3 |
Азот общий | См. ниже "Общее содержание азота" |
Азот органический | См. ниже "Общее содержание азота" |
Алюминий | Пробы берут в бутыли, промытые кислотой. Определение производят по возможности не позже чем через 2 ч после отбора пробы; прибавляют 5 мл HCl на 1 л пробы (в случае надобности) |
Аммиак | См. ниже "Ионы аммония и аммиак" |
Анионоактивные синтетические моющие вещества | Прибавляют 2 - 4 мл CHCl3 на 1 л пробы. Пробу анализируют не позже 1 суток после отбора |
Бораты | Обычно пробы не консервируют; их собирают в полиэтиленовую бутыль или в бутыль из стекла, из которого бор не выщелачивается |
БПК | Нельзя консервировать. Пробу хранят при 3 - 4 °C, обрабатывают не позже чем через 1 сутки |
Взвешенные вещества | Пробы не консервируют; определение следует производить не позже чем через 1 сутки. Хранение при температуре 3 - 4 °C |
Водородные ионы | См. "Кислотность" |
Вкус и привкус | Пробы нельзя консервировать; нельзя брать пробы в полиэтиленовые бутыли. Определение надо производить не позже чем через 2 ч после взятия пробы |
Гидроксильные ионы | См. "Кислотность" и "Щелочность" |
Гуминовые вещества | Пробы нельзя консервировать; их следует обработать не позже чем через 3 суток после взятия пробы |
Двуокись углерода | См. "Кислотность" и "Щелочность" |
Двуокись хлора | Пробы нельзя консервировать; определение надо производить сразу после взятия пробы |
Железо | Общее содержание железа: прибавляют 25 мл раствора HNO3 на 1 л пробы Различные формы железа: прибавляют 25 мл раствора ацетата натрия (68 г CH3COONa·3H2O в 500 мл H2O) и 25 мл раствора уксусной кислоты (166,7 мл CH3COOH (100%) в 500 мл H2O) на 1 л пробы При взятии пробы следует избегать соприкосновения воды с воздухом Внимание! Возможна адсорбция железа стенками сосуда для пробы |
Жесткость | Пробы не консервируют; см. "Кальций" и "Магний" |
Жирные кислоты | Пробы не консервируют; определение надо производить как можно раньше, в тот же день |
Жиры | См. "Экстрагируемые вещества" |
Запах | Пробы нельзя консервировать; определение производить не позже чем через 2 ч после взятия пробы, но не позднее конца дня отбора; некоторые запахи надо устанавливать сразу же на месте (запах хлорфенолов) |
Ионы аммония | Определение производят сразу же после отбора пробы, или пробу хранят при 3 - 4 °C, или прибавляют 1 мл H2SO4 на 1 л пробы, или прибавляют 2 - 4 мл CHCl3 на 1 л пробы |
Кадмий | Пробы надо собирать в чистые стеклянные или полиэтиленовые бутыли и прибавлять 5 мл HNO3 на 1 л пробы Внимание! Возможна адсорбция кадмия стенками бутыли |
Калий | Пробы надо собирать в полиэтиленовые сосуды или бутыли из стекла, из которого калий не выщелачивается Пробы не надо консервировать |
Кальций | Пробы обычно не консервируют |
Карбонаты | См. "Кислотность" и "Щелочность" |
Кислород | Пробы нельзя консервировать; их собирают в "кислородные" склянки, предназначенные для этой цели, при этом лучше всего использовать специальную насадку. Пробы надо фиксировать на месте прибавлением соответствующих реактивов |
Кислотность | Пробы нельзя консервировать. Определение производят сразу же на месте или производят отбор пробы при помощи специальной насадки (как при определении кислорода), сосуд заполняют пробой полностью, чтобы не осталось пузырьков воздуха; при транспортировке предохраняют пробу от нагревания и определение производят не позднее чем через 1 сутки |
Магний | Пробы обычно не консервируют |
Марганец | Определение производят сразу же после взятия пробы или прибавляют 5 мл HNO3 на 1 л пробы Внимание! Может произойти осаждение гидроокиси марганца, ее последующее окисление и адсорбция стенками сосуда |
Масла | См. "Экстрагируемые вещества" |
Медь | Прибавляют 5 мл HNO3 на 1 л пробы или прибавляют 5 - 10 мл HCl (1:1) на 1 л пробы Внимание! Возможна адсорбция меди стенками сосуда |
Минеральные масла | См. "Экстрагирующие вещества" |
Мышьяк | Определение производят не позже чем через 2 ч после отбора пробы; прибавляют 5 мл HCl на 1 л пробы; биологические процессы прекращают прибавлением 2 мл CHCl3 на 1 л пробы |
Мутность | Определение производят в тот же день или сохраняют пробу в темном месте и производят определение не позже, чем через 24 ч или прибавляют 2 - 4 мл CHCl3 на 1 л пробы (перед началом анализа пробу надо взболтать) |
Натрий | Пробы надо отбирать в полиэтиленовые бутыли или бутыли из стекла, из которого натрий не выщелачивается |
Никель | Прибавляют 5 мл HNO3 на 1 л пробы (нельзя консервировать при наличии цианидов) |
Нитраты | Определение производят в день взятия пробы или прибавляют 1 мл H2SO4 на 1 л пробы, или пробу охлаждают до 3 - 4 °C, или прибавляют 2 - 4 мл CHCl3 на 1 л пробы |
Нитриты | Определение производят сразу же после отбора пробы или прибавляют 1 мл H2SO4 на 1 л пробы, или пробу охлаждают до 3 - 4 °C, или прибавляют 2 - 4 мл CHCl3 на 1 л пробы |
Общее содержание азота | Определение производят не позже чем через сутки или прибавляют 1 мл H2SO4 на 1 л пробы, или прибавляют 2 - 4 мл CHCl3 на 1 л пробы |
Общее содержание фосфора | Пробы обычно не консервируют Внимание! Возможна адсорбция фосфатов стенками бутыли; нельзя консервировать кислотой (см. "Фосфаты") |
Озон | Пробы нельзя консервировать; определение надо производить сразу же после взятия пробы |
Окисляемость | а) По Кубелю: прибавляют 2 мл H2SO4 (1:2) на 100 мл пробы; при определении надо учитывать количество прибавленной кислоты б) Окисление бихроматом: прибавляют 1 мл H2SO4 на 1 л пробы в) Пробу надо охладить до 3 - 4 °C и провести определение не позже чем через 1 сутки |
Органический азот | См. "Общее содержание азота" |
pH | См. "Кислотность" |
Прозрачность | Пробы нельзя консервировать; определение производят сразу на месте взятия пробы, в крайнем случае не позже, чем через 1 сутки |
Растворенные вещества | Пробы не консервируют; определение надо производить не позже чем через сутки |
Свинец | Прибавляют 3 мл HNO3 на 1 л пробы или прибавляют 2 мл ледяной уксусной кислоты на 1 л пробы |
Серебро | Прибавляют 5 мл HNO3 на 1 л пробы Внимание! Возможна адсорбция серебра стенками бутыли |
Сероводород | См. "Сульфиды" |
Силикаты | Пробы собирают в полиэтиленовые бутыли, при высокой концентрации SiO2 прибавляют 1 мл H2SO4 (1:3) |
Смолы | См. "Экстрагируемые вещества" |
Содержание солей ("соленость") | Пробы нельзя консервировать |
Сульфаты | Пробы обычно не консервируют |
Сульфиды | Пробы надо собирать в отдельные бутыли, лучше всего с насадкой (как для определения кислорода); прибавляют 10 мл 10%-ного раствора ацетата кадмия или цинка на 1 л пробы Растворенные сульфиды: определение надо производить как можно раньше после отбора пробы, не консервируя ее |
Сульфиты | Определение производят как можно раньше после отбора пробы или пробу отбирают в отдельные бутыли, в которые предварительно вводят 0,2 мл 20%-ного раствора NaOH и 2 мл глицерина на 100 мл пробы |
Температура | Измерение производят сразу же на месте отбора проб |
Удельная электропроводность | Пробы нельзя консервировать; измерение надо производить не позже чем через 1 сутки |
Фенолы | Если содержание фенолов превышает 100 мг/л, определение надо производить, не консервируя пробы, не позже чем через 5 суток после отбора Пробы, содержащие менее 100 мг/л фенолов, можно консервировать добавлением 4 г NaOH на 1 л пробы Пробы, содержащие менее 0,05 мг/л фенолов, надо анализировать сразу после отбора |
Фосфаты | Определение производят как можно раньше после отбора пробы или прибавляют 2 - 4 мл CHCl3 на 1 л пробы и определение производят в тот же день Пробу нельзя консервировать кислотой |
Фториды | Пробы отбирают в полиэтиленовые бутыли, которые перед этим не были использованы для хранения в них проб с высоким содержанием фторидов |
Хлор | Пробы нельзя консервировать; их собирают в бутыли из темного стекла, предохраняя от действия солнечных лучей и от сотрясений Определение надо производить сразу же после отбора пробы (содержание хлора в пробе не является постоянным и при низкой температуре) |
Хлориды | Пробы обычно не консервируют. (В исключительных случаях прибавляют 2 - 4 мл CHCl3 на 1 л пробы, чтобы подавить биохимические процессы) |
Хром | Прибавляют 5 мл HNO3 на 1 л пробы Если надо раздельно определить Cr III и Cr VI, определение производят в тот же день Внимание! Возможна адсорбция хрома стенками бутыли |
Цианиды | Пробы, содержащие цианиды, при определении других компонентов нельзя консервировать кислотой Определение цианидов производят сразу же после отбора пробы Доводят значение pH пробы добавлением щелочи по крайней мере до 11 и сохраняют при температуре 3 - 4 °C |
Цинк | Прибавляют 1 мл H2SO4 на 1 л пробы (нельзя консервировать пробы при наличии в них цианидов) |
Щелочность | Пробы нельзя консервировать Определение производят сразу же на месте отбора проб Бутыль заполняют пробой доверху и приступают к анализу не позднее чем через 24 ч |
Экстрагируемые вещества | Пробу надо собирать в широкогорлую банку, в особенности при определении жиров и смолистых веществ Определение следует производить как можно раньше после отбора пробы При определении минеральных масел и жиров пробу консервируют добавлением 5 мл H2SO4 (1:1) на 1 л пробы Для консервирования нельзя пользоваться CHCl3 |
Данные о консервировании приведены при описании методов определения отдельных компонентов.
Транспортировка и хранение проб
1.14. Следует принимать все меры для того, чтобы сократить время между отбором пробы и ее анализом.
Наиболее благоприятным условием является расположение аналитической лаборатории недалеко от места отбора проб, лабораторий водопроводных станций, заводских лабораторий.
Транспортировать пробы следует быстро и осторожно. Желательно, чтобы проба была доставлена в лабораторию в день отбора. Не рекомендуется организовывать рабочие поездки на несколько дней, при которых пробы отбирают постепенно накапливают и лишь потом перевозят.
Для пересылки бутыли с пробами укладывают в ящики, имеющие перегородки. Каждую бутыль помещают в изолированное отделение. Промежутки между бутылями прокладывают эластичным материалом (бумагой, войлоком, резиной). Для пересылки по почте используются ящики с запирающимися крышками. Пробки бутылей следует тщательно укрепить. При морозной погоде стеклянные бутыли следует предохранять от замерзания в них пробы или пользоваться полиэтиленовыми бутылями. Определение большинства компонентов допускает транспортировку проб в лабораторию.
Если пробы для определения отдельных компонентов нельзя транспортировать кратчайшим путем с соблюдением всех мер предосторожности, описанных выше, применяют консервирование или фиксацию проб на месте. Способы консервирования описаны при изложении отдельных методов, а перечень их приведен в табл. 1 (стр. 13 - 20).
Следует помнить, что ни консервирование, ни фиксация не обеспечивают постоянного состава пробы на неограниченное время. Целью этих мероприятий является лишь сохранение соответствующего компонента без изменений на время перевозки. К анализу надо приступать в кратчайший срок.
До начала анализа пробы сохраняют в холодильнике и вынимают их только перед самым началом работы. Приступают к анализу, когда температура воды сравняется с комнатной температурой. С целью получения точных результатов анализа требуется строго соблюдать установленное время хранения. Пробы, взятые не специалистами, неточно обозначенные и доставленные в лабораторию через несколько дней после отбора, бесполезны, и анализ их делать бессмысленно, так как получаемые результаты ненадежны.
Температура
1.15. Измерение температуры воды и температуры воздуха во время отбора пробы является неотделимой частью анализа.
Температура воды измеряется одновременно с отбором пробы ртутным термометром с ценой деления 0,1 - 0,5 °C.
Там, где позволяют местные условия, температуру поверхностных вод измеряют погружением термометра в воду (прямой солнечный свет необходимо затенить). Если непосредственное измерение в водоеме выполнить невозможно, то температуру измеряют в бутыли сразу же после отбора пробы. Температура бутыли емкостью не менее 1 л перед отбором пробы должна быть приведена к температуре воды погружением в исследуемую воду и при измерении температуры она не должна подвергаться влиянию каких-либо источников тепла или действию прямого солнечного света. При отборе пробы прямо из крана температура измеряется в струе.
В большинстве случаев температуру отсчитывают после установления на неизменном уровне ртутного столбика термометра, погруженного в исследуемую воду. Только при измерении температуры проб, температура которых значительно отличается от окружающей среды (например, вода из родников, из глубины водохранилища), не выжидают установления столбика ртути на постоянном уровне, а записывают наивысшее показание термометра при измерении пробы, температура которой выше температуры окружающей среды, и самое низкое показание термометра при измерении пробы, температура которой ниже температуры окружающей среды.
Для измерения температуры воды водоема на различных глубинах можно применить также специальные термометры. Способ работы ими зависит от типа применяемого прибора.
Температуру воздуха измеряют сухим термометром в тени вне воздействия какого-либо источника тепла на высоте 1 м от поверхности земли в течение времени, необходимого для установки ртутного столбика термометра на постоянном уровне.
Температуры воздуха и воды приводят в градусах Цельсия с округлением до 0,1 или 0,5° в зависимости от типа применяемого термометра. Знак ставится только при температурах ниже нуля.
Общие положения
2.1. Прозрачность зависит от цвета воды и ее мутности. Мерой прозрачности служит высота водяного столба, сквозь который можно еще наблюдать черный крест на белой доске с крестом из черных линий шириной 1 мм определенных размеров или прочесть шрифт определенного типа. Прозрачность воды характеризует наличие в ней взвешенных и коллоидных примесей.
Определение прозрачности производится в поверхностных водах на месте отбора пробы и при оценке работы водоочистных станций.
Для определения прозрачности применяют два метода: по "кресту" и по стандартному шрифту. Определение по кресту применяют при контроле работы очистных сооружений водопроводов и качества воды в водопроводной сети; в остальных случаях применяют определение прозрачности по "шрифту".
Пробы для определения прозрачности нельзя консервировать. Определение производят сразу на месте взятия пробы, в крайнем случае не позже чем через сутки; для одного определения прозрачности по кресту требуется 2 л воды, для одного определения прозрачности по шрифту требуется 0,5 л воды.
Определение прозрачности воды по "кресту"
(основной метод)
Аппаратура
2.2. Прибор для определения прозрачности по кресту (рис. 1) состоит из стеклянной трубы диаметром 3 см, длиной 350 см, градуированной по высоте на сантиметры. Нижний конец трубы закрыт резиновой пробкой, снабженной спускным отверстием с зажимом. На дне трубы, на пробке, помещается белый фарфоровый диск с черными линиями, образующими крест из линий шириной в 1 мм, и с четырьмя черными точками диаметром в 1 мм по одной в середине каждой образованной линиями четверти диска.

1 - стеклянный цилиндр; 2 - шкала; 3 - тяга зажима;
4 - зажим с грузом; 5 - резиновая пробка; 6 - фарфоровый
диск с черными линиями и точками; 7 - кожух электрической
лампы; 8 - резиновый шланг
Определение производят при искусственном освещении, для чего применяют электрическую лампу 300 Вт, установленную у дна трубы.
Ход определения
2.3. Трубу наполняют исследуемой водой до полного исчезновения креста. Затем, после выделения пузырьков воздуха, воду постепенно спускают до появления в поле зрения отчетливо видимых точек. Глаз наблюдателя располагается на высоте около 5 см над концом трубы.
Толщина слоя воды, выраженная в сантиметрах, отвечающая моменту видимости точек креста, характеризует прозрачность воды по кресту.
По окончании определения трубу опорожняют и споласкивают чистой водой.
Для приблизительного перевода величины прозрачности по кресту к величине мутности служит табл. 2.
Таблица 2
Прозрачность, см | Мутность, мг·л | Прозрачность, см | Мутность, мг·л | Прозрачность, см | Мутность, мг·л | Прозрачность, см | Мутность, мг·л | Прозрачность, см | Мутность, мг·л |
3,5 | 270 | 27 | 33,8 | 62 | 14,8 | 108 | 8,3 | 270 | 3,45 |
4 | 235 | 28 | 32,6 | 63 | 14,6 | 110 | 8,2 | 280 | 3,5 |
4,5 | 205 | 29 | 31,5 | 64 | 14,4 | 112 | 8,1 | 290 | 3,2 |
5 | 185 | 30 | 30,5 | 65 | 14,2 | 114 | 8,0 | 300 | 3,1 |
5,5 | 170 | 31 | 29,5 | 66 | 14 | 116 | 7,9 | 310 | 3 |
6 | 155 | 32 | 28,6 | 67 | 13,8 | 118 | 7,75 | ||
6,5 | 142 | 33 | 27,7 | 68 | 13,6 | 120 | 7,65 | ||
7 | 130 | 34 | 26,9 | 69 | 13,4 | 122 | 7,55 | ||
7,5 | 122 | 35 | 26,1 | 70 | 13,2 | 124 | 7,45 | ||
8 | 114 | 36 | 25,4 | 71 | 13 | 126 | 7,35 | ||
8,5 | 102 | 37 | 24,8 | 72 | 12,8 | 128 | 7,25 | ||
9 | 102 | 38 | 24,2 | 73 | 12,6 | 130 | 7,15 | ||
9,5 | 97 | 39 | 23,6 | 74 | 12,4 | 132 | 7,05 | ||
10 | 92 | 40 | 23 | 75 | 12,2 | 134 | 6,9 | ||
10,5 | 87 | 41 | 22,4 | 76 | 12,05 | 136 | 6,8 | ||
11 | 83 | 42 | 21,8 | 77 | 11,9 | 138 | 6,7 | ||
11,5 | 79 | 43 | 21,2 | 78 | 11,75 | 140 | 6,6 | ||
12 | 76 | 44 | 20,7 | 79 | 11,6 | 145 | 6,3 | ||
12,5 | 73 | 45 | 20,2 | 80 | 11,45 | 150 | 6,1 | ||
13 | 70 | 46 | 19,7 | 81 | 11,3 | 155 | 5,9 | ||
13,5 | 67,5 | 47 | 19,3 | 82 | 11,05 | 160 | 5,75 | ||
14 | 65 | 48 | 18,9 | 83 | 11 | 165 | 5,6 | ||
14,5 | 63 | 49 | 18,5 | 84 | 10,85 | 170 | 5,45 | ||
15 | 61 | 50 | 18,4 | 85 | 10,7 | 175 | 5,3 | ||
16 | 56,4 | 51 | 17,9 | 86 | 10,35 | 180 | 5,15 | ||
17 | 53,1 | 52 | 17,6 | 89 | 10,3 | 185 | 5,0 | ||
18 | 50,4 | 53 | 17,3 | 90 | 10,1 | 190 | 4,85 | ||
19 | 48 | 54 | 17 | 92 | 9,9 | 195 | 4,75 | ||
20 | 45,5 | 55 | 16,7 | 94 | 9,7 | 200 | 4,6 | ||
21 | 43,3 | 56 | 16,4 | 96 | 9,5 | 210 | 4,4 | ||
22 | 41,4 | 57 | 16,1 | 98 | 9,3 | 220 | 4,2 | ||
23 | 39,6 | 58 | 15,8 | 100 | 9,1 | 230 | 4,0 | ||
24 | 38 | 59 | 15,5 | 102 | 8,9 | 240 | 3,85 | ||
25 | 36,5 | 60 | 15,2 | 104 | 8,7 | 250 | 3,7 | ||
26 | 35,1 | 61 | 15 | 106 | 8,5 | 260 | 3,55 |
Определение прозрачности при помощи шрифта (по Снеллену)
2.4. Определяется высота водяного столба, при которой стандартный шрифт, находящийся под дном измерительного цилиндра, становится плохо различимым. Образец шрифта см. приложение 1.
Аппаратура
2.5. Измерительный цилиндр Снеллена (рис. 2) - стеклянный цилиндр с плоским дном, высотой 30 - 35 см, диаметром 2,5 - 3 см. Цилиндр градуирован на сантиметры на высоту 30 см от дна. В нижней части цилиндра имеется отводная трубка с надетой на нее каучуковой трубкой с зажимом, через которую выпускают воду из цилиндра для уменьшения высоты ее столба.

при помощи шрифта
Ход определения
2.6. Испытуемую воду перед определением хорошо взбалтывают и наливают в цилиндр на высоту, предположительно отвечающую прозрачности воды, затем ставят цилиндр неподвижно над шрифтом <1> так, чтобы он находился в 4 см от дна. Добавляя или отливая воду из цилиндра, находят предельную высоту столба воды, при которой чтение шрифта еще возможно.
--------------------------------
<1> Образец шрифта - см. приложение.
Определение производят в хорошо освещенном помещении, не на прямом солнечном свету, на расстоянии 1 м от окна.
Прозрачность выражают в сантиметрах высоты столба воды с точностью до 0,5 см.
По окончании определения цилиндр опорожняют и споласкивают чистой водой.
Общие положения
3.1. Под взвешенными веществами понимаются содержащиеся в воде частицы минерального и органического происхождения, имеющие размер более 1·10-4 мм.
Определение концентрации взвешенных в воде веществ производится фотометрически (основной метод для вод, содержащих до 100 мг/л взвешенных веществ) или весовым методом с мембранными (до 100 мг/л) или бумажными фильтрами (более 100 мг/л).
Пробы воды для определения концентрации взвешенных веществ не консервируют, их отбирают в бутыли из стекла или полиэтилена, определение производят не позднее чем через сутки. Объем пробы 500 мл при содержании взвешенных веществ более 50 мг/л и 1000 мл при меньшем их содержании.
Результаты определения выражаются в миллиграммах на 1 л воды.
Фотометрический метод определения содержания
взвешенных веществ
3.2. Принцип метода. На фотометре определяют коэффициент светопропускания образца воды и по калибровочной кривой устанавливают концентрацию в ней взвешенных веществ, соответствующую данному коэффициенту светопропускания.
Аппаратура. Реактивы. Калибровочная кривая
3.3. Фотоколориметр. При исследовании природных вод 100 г глины, отобранной вблизи водоисточника, растирают с дистиллированной водой в фарфоровой ступке, смывая растертую глину в стеклянный сосуд диаметром 20 - 25 см и высотой 30 - 50 см. Затем сосуд дополняют дистиллированной водой доверху и, тщательно перемешав его содержимое, оставляют в покое на 60 мин. По прошествии этого времени сифоном отбирают из сосуда верхний слой воды на глубину 180 мм. Этот слой содержит глинистую взвесь с частицами, имеющими гидравлическую крупность менее 0,05 мм/сек. Взвесь из воды отфильтровывают на плотном бумажном фильтре, высушивают в сушильном шкафу при 105 °C и растирают в агатовой ступке. Отвесив 1 г взвеси, ее снова растирают в ступке с дистиллированной водой, смывая в мерную колбу емкостью 1 л, в которую предварительно было налито 200 мл 0,1%-ного раствора гексаметафосфата натрия (стабилизатор). Объем суспензии в мерной колбе доводят дистиллированной водой до метки; эта суспензия содержит 1 мг взвеси в 1 мл. Из нее разбавлением готовят суспензии с содержанием взвешенных веществ 1, 2, 5, 10, 20, 40, 60 мг/л, пользуясь которыми строят калибровочную кривую фотометра, применяя для малых концентраций (до 10 мг/л) кювету длиной 50 мм, и меньшие кюветы для образцов с  содержанием взвешенных веществ.
содержанием взвешенных веществ.
содержанием взвешенных веществ.При исследовании воды в процессе ее осветления на водопроводных очистных сооружениях калибровочную кривую получают так же, как при исследовании природных вод, но в качестве эталонного замутнителя вместо глины применяют осадок из отстойника, осветлителя или промывной воды фильтра.
Ход определения
3.4. В правый и левый пучки света фотометра устанавливают кюветы с исследуемой водой, из которой предварительно центрифугированием или фильтрованием удалены взвешенные вещества. Стрелку гальванометра устанавливают на нуль и затем, заменив правую кювету с водой, не содержащей взвеси, кюветой с образцом исследуемой воды, определяют ее коэффициент светопропускания, по которому с помощью калибровочной кривой определяют содержание в исследуемой воде взвешенных веществ.
Чувствительность метода 0,2 мг/л, точность 5%.
Определение при помощи мембранных фильтров
Аппаратура
3.5. Фильтровальный аппарат Зейтца (или Олихова) с колбой для отсасывания, предохранительной склянкой и водоструйным насосом (рис. 3).

1 - стеклянный цилиндр; 2 - гайка с резьбой; 3 - кран;
4 - вакуумный шланг; 5 - резиновая пробка
Мембранные фильтры (предварительные или N 4). Мембранные фильтры кипятят 15 мин в дистиллированной воде. Кипячение повторяют 3 раза, каждый раз в свежей дистиллированной воде. Затем фильтры просушивают при 105 °C до постоянного веса (на что требуется 2 - 3 ч) и взвешивают. Перед взвешиванием фильтры нумеруют карандашом, веса фильтров заносят в журнал.
Сушильный шкаф (105 °C).
Ход определения
3.6. Предварительно взвешенный фильтр закрепляют в аппарате Зейтца. В зависимости от количества взвешенных веществ пробу объемом 100 - 500 мл фильтруют под вакуумом. Частички, приставшие к стенкам аппарата, смывают дистиллированной водой на фильтр.
После фильтрования аппарат разбирают. Фильтр подсушивают на воздухе, затем - в сушильном шкафу при 105 °C до постоянного веса и взвешивают.
Расчет. Общее содержание взвешенных веществ (x) в мг/л вычисляют по формуле
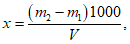
где m2 - вес мембранного фильтра с осадком, мг;
m1 - вес мембранного фильтра до работы, мг;
V - объем пробы, взятой для определения, мл.
Результаты округляются до целых миллиграммов, а при значениях выше 1000 мг/л - до 10 мг.
Определение при помощи бумажных фильтров
Аппаратура
3.7. Бумажные фильтры (синяя лента). Для количественного определения фильтр высушивают около 1 ч при 105 °C до постоянного веса и взвешивают в бюксе.
Сушильный шкаф (105 °C).
Ход определения
3.8. Объем воды, необходимый для определения, зависит от предполагаемого содержания в ней взвешенных веществ; он может быть найден по следующим данным:
Предполагаемое содержание взвешенных веществ, мг/л | Менее 10 | 10 - 50 | 50 - 100 | 100 - 500 | Более 500 |
Объем пробы, л | 1,5 - 2 | 1,0 | 0,5 | 0,25 | 0,1 |
Пробу тщательно взбалтывают и быстро, не давая осесть взвеси, отбирают мерным цилиндром необходимое для определения количество воды (согласно приведенным данным) и фильтруют через подготовленные бумажные фильтры. Если фильтрат мутный, его следует профильтровать вторично через тот же фильтр. Взвешенные вещества на фильтре промывают небольшим количеством холодной воды. Дав воде полностью стечь, помещают фильтр в предварительно взвешенный бюкс и высушивают при 105 °C до постоянного веса.
После охлаждения в эксикаторе бюкс с фильтром взвешивают.
Расчет. Содержание взвешенных веществ (x) в мг/л вычисляют по формуле

где m1 - вес бюкса с высушенным бумажным фильтром, мг;
m2 - вес бюкса с фильтром и взвешенными веществами после высушивания, мг;
V - объем пробы, взятой для определения, мл.
Результаты округляются до целых миллиграммов, а значения, превышающие 1000 мг/л, - до 10 мг.
Общие положения
4.1. Причиной окраски различных природных вод обычно являются вещества, извлекаемые водой из торфа, гумуса, болотной почвы, отмерших растений. Допустимая цветность питьевой воды по шкале в градусах не более 20 (ГОСТ 2874-54). У вод, содержащих большое количество взвешенных веществ, цвет определяется после отстаивания. Поэтому объективно определить цвет проб довольно трудно. Если объективное определение цвета провести нельзя, оттенок и интенсивность цвета описываются словесно.
Основными методами определения цветности являются:
1) визуальное сравнение со стандартными растворами, содержащими бихромат калия и соль кобальта, и 2) сравнение с теми же стандартными растворами на фотоколориметре (основной метод).
При определении цветности пробы не консервируют. Определение проводят через 2 ч после отбора пробы.
Метод сравнения со стандартными растворами бихромата
калия и сульфата кобальта
4.2. Пробы воды, отвечающие по своей окраске цветам смеси растворов бихромата калия и сульфата кобальта, сравниваются со стандартными растворами, концентрации которых известны.
Для определения необходима проба воды объемом не менее 500 мл.
Мешающие влияния
4.3. Определению мешает мутность воды, поэтому определение цветности производится после фильтрования пробы через стеклянную фильтрующую пластинку N 4 или центрифугирования. В этом случае при записи надо указать использованный способ обработки пробы. Если прозрачность ниже 20 см по Снеллену (см. стр. 22), то воду перед определением центрифугируют.
Аппаратура
4.4. Колориметрические цилиндры Несслера емкостью 100 мл из бесцветного стекла с плоским дном.
Реактивы
4.5. Калия бихромат
Кобальта сульфат
Кислота серная
Калибровочная кривая
4.6. Растворяют отдельно в дистиллированной воде 0,0875 г бихромата калия (K2Cr2O7) и 2,000 г сульфата кобальта (CoSO4·7H2O). Смешивают оба раствора, прибавляют 1 мл химически чистой серной кислоты (плотность 1,84) и доводят дистиллированной водой до 1 л. Этот раствор имеет цветность 500° (раствор N 1).
Для приготовления раствора N 2 берут 1 мл серной кислоты и доводят дистиллированной водой до 1 л (в мерной колбе).
Смешением растворов N 1 и N 2 в соотношениях, приведенных ниже, получают шкалу цветности в градусах цветности, которую сохраняют в темноте в колориметрических цилиндрах Несслера, закрытых резиновыми пробками.
Раствор N 1, мл | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 8 | 10 | 12 | 14 | 16 |
Раствор N 2, мл | 100 | 99 | 98 | 97 | 96 | 95 | 94 | 92 | 90 | 88 | 86 | 84 |
Цветность, град | 0 | 5 | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 60 | 70 | 80 |
Ход определения
4.7. Если прозрачность исходной воды меньше 20 см по шрифту Снеллена, пробу фильтруют. В колориметрический цилиндр Несслера, одинаковый с теми, из которых приготовлена шкала, наливают 100 мл испытуемой воды и, просматривая сверху на белом фоне, находят цилиндр шкалы, окраска в котором совпадает с окраской испытуемой воды.
Если анализируемая вода имеет цветность больше 80°, то определение производят после разбавления ее дистиллированной водой. Величина цветности в этом случае получается умножением результата определения на кратность разбавления.
Точность метода
Результаты определения выражают в градусах цветности со следующей точностью:
Цветность, град | 1 - 50 | 51 - 100 | 101 - 250 | 251 - 500 |
Точность, град | 2 | 5 | 10 | 20 |
Метод сравнения со стандартными растворами бихромата калия
и сульфата кобальта на фотоколориметре
Аппаратура
4.8. При определении цветности воды желательно пользоваться фотометром (фотоколориметром).
Фотоколориметры, прокалиброванные по набору стандартных растворов различных концентраций, темно-фиолетовый светофильтр (N 1 для ФЭКН-57, N 2 для ФЭКН-56).
Кюветы длиной 50 мм.
Реактивы
4.9. Стандартные растворы для калибровки прибора (см. стр. 29). В левую кювету наливают дистиллированную воду. В правую кювету последовательно наливают стандартные растворы различной концентрации. Для каждого стандартного раствора определяют коэффициент светопропускания.
Откладывая по оси абсцисс цветность каждого раствора, а по оси ординат - соответствующий ему коэффициент светопропускания, строят калибровочную кривую.
Ход определения
4.10. В правый пучок света помещают кювету с исследуемым раствором, а в левый - кювету с дистиллированной водой. Индекс левого барабана устанавливают на нулевое деление шкалы оптической плотности. Вращением круговых фотометрических клиньев стрелку гальванометра устанавливают на нуль, рукоятка чувствительности находится в положении 1, ставят ее в положение 2, проводят точную установку стрелки.
Затем рукоятку чувствительности снова ставят в положение 1 и в правый пучок света вводят кювету с дистиллированной водой; при этом стрелка гальванометра отклоняется от нулевого положения. Вращением измерительных барабанов стрелку гальванометра вновь устанавливают на нуль, сначала при малой (положение 1), а затем при максимальной чувствительности (положение 2) прибора. Величину коэффициента светопропускания отсчитывают по левому барабану.
По калибровочной кривой определяют цветность, соответствующую данному коэффициенту светопропускания.
Если цветность исходной воды больше 80°, определение производят после разбавления дистиллированной водой. Величину цветности в этом случае получают умножением результата определения на кратность разбавления.
Общие положения
5.1. Вкусовые свойства воды зависят от присутствия в ней веществ природного происхождения или веществ, которые попадают в воду в результате загрязнения воды сточными водами, ядохимикатами.
5.2. Вкус и привкус воды определяют органолептически качественно и количественно (по интенсивности). Различают четыре вида вкуса: соленый, горький, сладкий, кислый. Остальные виды вкусовых ощущений называются привкусами.
Вкус и привкус определяют в сырой воде, за исключением воды открытых водоемов и источников, сомнительных в санитарном отношении, вкус воды которых определяют после кипячения и охлаждения до комнатной температуры, что должно быть отмечено в записи анализа.
Вкус и привкус воды, подвергаемой хлорированию, определяют через 30 мин после введения хлора.
Результат определения зависит от вкусового восприятия и опытности исследователя. Во избежание ошибки необходимо, чтобы определение привкуса проводило несколько человек. При определении привкуса питьевой воды 15 - 20 мл ее набирают в рот и держат несколько секунд; проглатывать ее не рекомендуется.
Качественную характеристику привкуса определяют по соответствующим признакам и выражают описательно: хлорный, рыбный, металлический.
Интенсивность вкуса и привкуса определяют по пятибалльной системе (аналогично определению запаха, см. табл. 4).
Общие положения
6.1. Запах воды вызывают летучие пахнущие вещества, попадающие в нее естественным путем или со сточными водами. Из неорганических веществ запах может давать только сероводород.
Характер и интенсивность запаха определяют органолептически.
Запахи воды по характеру разделяют на две группы:
а) запахи естественного происхождения (образующиеся в результате жизнедеятельности и отмирания водных организмов, от естественных природных стоков);
б) запахи искусственного происхождения (от промышленных сточных вод, гербицидов, инсектицидов).
Определение характера запаха
6.2. Анализируемую воду, температура которой 15 - 20 °C, наливают в коническую колбу емкостью 300 мл с широким горлом на 2/3 ее объема, накрывают часовым стеклом, взбалтывают содержимое вращательным движением, открывают и втягивают носом воздух из колбы. Характер запаха выражают описательно. Для запахов первой группы (естественного происхождения) дают определение по классификации, приведенной в табл. 3.
Таблица 3
Символ | Характер запаха | Примерный род запаха |
А | Ароматический | Огуречный, цветочный |
Б | Болотный | Илистый, тинистый |
Г | Гнилостный | Фекальный, сточный |
Д | Древесный | Запах мокрой щепы, древесной коры |
З | Землистый | Прелый, свежевспаханной земли, глинистый |
П | Плесневый | Затхлый, застойный |
Р | Рыбный | Рыбьего жира, рыбы |
С | Сероводородный | Тухлых яиц |
Т | Травянистый | Скошенной травы, сена |
Н | Неопределенный | Запахи естественного происхождения, не подходящие под предыдущие определения |
Запахи второй группы называют по соответствующим веществам: фенольный, хлорфенольный, бензинный, хлорный.
Запах воды, подвергаемой хлорированию, определяется через 30 мин после введения хлора.
Определение интенсивности запаха
6.3. Определение интенсивности запаха производят органолептически по балльной системе и по методу порогового разбавления.
Балльные оценки запаха производят при температуре 15 - 20 °C и при нагревании воды до 60 °C.
Нагревание производят в конической колбе, закрытой часовым стеклом. После нагревания жидкости до 60 °C содержимое колбы перемешивают вращательным движением, снимают часовое стекло и втягивают носом воздух из колбы.
Запах оценивают по табл. 4, интенсивность его выражают в баллах.
Таблица 4
Интенсивность запаха, балл | Характеристика | Описательные определения |
0 | Никакого | Отсутствие ощутимого запаха |
1 | Очень слабый | Запах, не замечаемый потребителем, но обнаруживаемый в лаборатории опытным исследователем |
2 | Слабый | Запах, не привлекающий внимания потребителя, но обнаруживаемый им, если указать на него |
3 | Заметный | Запах, легко обнаруживаемый и могущий дать повод относиться к воде с неодобрением |
4 | Отчетливый | Запах, обращающий на себя внимание и делающий воду неприятной для питья |
5 | Очень сильный | Запах настолько сильный, что делает воду непригодной для питья |
Определение запаха зависит от опытности и индивидуальных способностей исследователя. Для исключения субъективной ошибки целесообразно определение запаха производить группе из 3 - 5 человек.
При проведении работы по определению запаха должны соблюдаться следующие условия:
а) помещение, в котором производится определение запаха, должно быть чистым, без запаха;
б) должно быть обеспечено отсутствие какого-либо запаха от рук, платья наблюдателя;
в) одному и тому же лицу нельзя производить определение запаха длительное время, так как наступает утомляемость, привыкание.
Метод порогового разбавления
6.4. Исследуемую воду разбавляют специально подготовленной непахнущей дистиллированной водой до такой степени, что запах нагретой до 60 °C смеси становится едва заметным ("пороговый запах"). Необходимая для этого кратность разбавления является количественной оценкой интенсивности запаха исследуемой воды.
Кратность разбавления называют "пороговым числом". Чем больше пороговое число, тем интенсивнее запах исходной воды.
Аппаратура
6.5. Конические колбы емкостью 500 мл с притертыми пробками.
Водяная баня.
Трубка с активным углем. Стеклянную трубку диаметром 20 - 30 мм с коническим нижним концом заполняют на высоту 700 - 800 мм активным углем марки "БАУ" (ГОСТ 6217-52) с крупностью зерен 1 - 3,5 мм. Нижнюю часть трубки на высоту 20 - 30 мм заполняют стеклянной ватой. Перед загрузкой в трубку активный уголь кипятят в дистиллированной воде в течение 2 - 3 ч, дважды сменяя воду.
Реактивы
6.6. Через подготовленную стеклянную трубку фильтруют дистиллированную воду, имеющую комнатную температуру, со скоростью не более 5 м/час. Фильтрат нагревают в колбе до 60 °C и проверяют на отсутствие запаха.
Ход определения
6.7. Для определения интенсивности запаха в конические колбы берут различные количества исследуемой воды (см. табл. 5), доводят объем воды в каждой колбе до 200 мл непахнущей дистиллированной водой, закрывают колбы часовыми стеклами и нагревают на плитке до 60 °C.
Таблица 5
Объем анализируемой воды, мл | Объем дистиллированной воды для разбавления, мл | Интенсивность запаха, "пороговое число" | Объем анализируемой воды, мл | Объем дистиллированной воды для разбавления, мл | Интенсивность запаха, "пороговое число" |
200 | - | 1 | 12 | 118 | 17 |
140 | 60 | 1,4 | 8,3 | 192 | 24 |
100 | 100 | 2 | 5,7 | 194 | 35 |
70 | 130 | 3 | 4 | 196 | 50 |
50 | 150 | 4 | 2,8 | 197 | 70 |
35 | 165 | 6 | 2 | 198 | 100 |
25 | 175 | 8 | 1,4 | 200 | 140 |
17 | 183 | 12 | 1 | 199 | 200 |
Одновременно в такую же колбу наливают для сравнения 200 мл непахнущей дистиллированной воды и также нагревают до 60 °C.
Сначала определяют наличие запаха в контрольной пробе с непахнущей дистиллированной водой, для чего содержимое колбы слегка взбалтывают круговыми движениями, приподнимают часовое стекло и энергично втягивают носом воздух из колбы.
Затем аналогичным образом определяют запах в колбе с наибольшим разбавлением исходной воды (с наибольшим "пороговым числом").
Если запах не ощущается, то переходят к колбе со следующим, меньшим "пороговым числом" до тех пор, пока не будет установлен едва заметный запах. Интенсивность запаха исходной воды будет равна "пороговому числу" этой последней колбы.
Если в первой же колбе с наибольшим "пороговым числом" при проведении опыта будет обнаружен запах, опыт нужно повторить с большими разбавлениями исходной воды.
Расчет. Пороговую интенсивность запаха (P) определяют по табл. 5 или по формуле

где a - объем пробы, взятой для получения смеси с ощутимым запахом, мл.
Общие положения
7.1. Кислотность природных вод с pH более 4,5 зависит в основном от содержания свободной двуокиси углерода и в некоторых случаях от присутствия гуминовых и других слабых органических кислот. Если pH воды менее 4,5 (например, водород-катионированная вода), в ней содержатся также сильные кислоты и соли сильных кислот и слабых оснований.
Различают следующие виды кислотности:
а) общая кислотность - эквивалентна расходу сильного основания (например, NaOH) на реакцию с сильными и слабыми кислотами (включая CO2) при доведении pH раствора до 8,3;
б) свободная кислотность - эквивалентна расходу сильного основания на реакцию только с сильными кислотами при доведении pH от величины менее 4,5 до pH = 4,5;
в) кислотность, зависящая от слабых нелетучих кислот (гуминовых и др.). Концентрация их эквивалентна расходу сильного основания на титрование пробы воды после удаления из нее свободной двуокиси углерода, от pH 4,5 до pH 8,3 (или от перехода окраски метилового оранжевого до перехода окраски фенолфталеина);
г) свободная двуокись углерода, содержание ее равно общей кислотности, за вычетом свободной кислотности и кислотности, зависящей от свободных гуминовых и других нелетучих кислот (при наличии в воде свободного сероводорода вычитается также его концентрация);
д) агрессивная двуокись углерода - часть свободной двуокиси углерода, способная растворять карбонат кальция.
Определение всех видов кислотности производят титрованием проб воды растворами сильных оснований.
Содержание в воде свободной и агрессивной углекислоты можно определять по номограммам на основе данных химических анализов воды.
Аппаратура
7.2. Бюретки для титрованных растворов едкого натра.
pH-метр со стеклянным и каломельным электродами.
Реактивы
7.3. Едкий натр, 15 н. раствор (запасный): растворяют 625 г NaOH ч.д.а. в 800 мл дистиллированной воды и оставляют для осаждения карбоната натрия не менее чем на 48 ч. Хранят в плотно закрытом полиэтиленовом сосуде.
Едкий натр, 0,1 н. раствор (рабочий): 6,6 мл 15 н. запасного раствора NaOH доводят до 1 л прокипяченной дистиллированной водой. Раствор хранят в сосуде, закрытом пробкой с хлоркальциевой трубкой, наполненной натронной известью. Годность раствора - не более 1 недели.
Поправочный коэффициент устанавливают по 0,1 н. титрованному раствору HCl.
Едкий натр, 0,02 н. (рабочий): 200 мл 0,1 н. раствора NaOH доводят прокипяченной дистиллированной водой до 1 л. Хранят так же, как 0,1 н. раствор NaOH.
Годность раствора - не более 1 недели.
Поправочный коэффициент устанавливают по 0,02 н. раствору HCl.
Метиловый оранжевый, 0,05%-ный водный раствор. Растворяют 0,5 г метилового оранжевого в 1 л дистиллированной воды.
Фенолфталеин, 0,5%-ный спиртовой раствор: 0,5 г фенолфталеина растворяют в 50 мл 96%-ного этилового спирта и добавляют 50 мл дистиллированной воды. В раствор по каплям добавляют 0,02 н. раствор NaOH до появления слабой розовой окраски.
Сегнетовая соль, 50%-ный раствор. Растворяют 50 г кристаллической сегнетовой соли (калий-натрий виннокислый) в 100 мл прокипяченной дистиллированной воды, дают отстояться несколько дней, нейтрализуют 0,1 н. раствором NaOH по фенолфталеину и фильтруют через стеклянную вату. Хранят в склянке темного стекла.
Определение свободной кислотности
7.4. К 100 мл исследуемой воды добавляют 3 капли раствора метилового оранжевого и при появлении красного окрашивания титруют 0,1 н. раствором NaOH до перехода окраски в золотисто-розовую. Титрование можно проводить без индикатора, контролируя повышение величины pH раствора. Заканчивается титрование при pH = 4,5. Если при добавлении индикатора к воде раствор становится желтым, свободная кислотность равна нулю.
Расчет. Свободную кислотность Kсв в мг-экв/л вычисляют по формуле
Kсв = ka,
где k - поправочный коэффициент для приведения концентрации раствора NaOH к точно 0,1 н.;
a - расход 0,1 н. раствора NaOH на титрование, мл.
Определение общей кислотности
7.5. К 100 мл исследуемой воды добавляют 5 - 10 капель раствора фенолфталеина и титруют 0,1 н. раствором NaOH до появления слабо-розового окрашивания, не исчезающего в течение 2 - 3 мин. Титрование можно проводить без индикатора до получения устойчивого значения pH = 8,3.
Расчет. Общую кислотность Kоб в мг-экв/л вычисляют по формуле
Kоб = kb,
где k - поправочный коэффициент для приведения концентрации раствора NaOH к точно 0,1 н.;
b - расход 0,1 н. раствора NaOH на титрование, мл.
Определение кислотности, обусловленной
слабыми нелетучими кислотами
7.6. Нейтральную по метиловому оранжевому пробу воды объемом 100 мл (например, после определения свободной кислотности) кипятят в течение 2 мин для удаления свободной двуокиси углерода, быстро охлаждают, добавляют 5 - 10 капель индикатора фенолфталеина и титруют 0,1 н. раствором NaOH до появления устойчивого слабо-розового окрашивания.
Расчет. Кислотность, зависящую от присутствия в воде слабых нелетучих кислот Kсл в мг-экв/л, вычисляют по формуле
Kсл = kc,
где c - расход 0,1 н. раствора NaOH на титрование, мл.
Определение концентрации свободной двуокиси
углерода титрованием
7.7. Мерную колбу емкостью 100 мл с узким длинным горлом наполняют исследуемой водой точно до метки, прибавляют 5 - 10 капель индикатора фенолфталеина и титруют 0,1 н. раствором NaOH. Титрование проводят, добавляя раствор NaOH небольшими порциями (к концу по 1 капле), каждый раз закрывают колбу резиновой пробкой и перемешивают содержимое колбы плавным покачиванием. Титрование заканчивают при появлении устойчивой в течение 2 - 3 мин розовой окраски.
При необходимости точного анализа определение повторяют, при этом к пробе воды сразу добавляют весь объем раствора NaOH, определенный по первому титрованию (в случае расхода 0,1 н. раствора NaOH более 1 мл), либо большую его часть (если при первом титровании расход 0,1 н. раствора NaOH составлял менее 1 мл), затем добавляют 5 - 10 капель фенолфталеина и дотитровывают по каплям до устойчивого розового окрашивания. Расчет проводят по результату второго титрования.
Расчет. Содержание в воде свободной двуокиси углерода [CO2]св в мг/л вычисляют по формуле
[CO2]св = 44(kd - Kсв - Kсл),
где k - поправочный коэффициент для приведения концентрации раствора NaOH к точно 0,1 н.;
d - расход 0,1 н. раствора NaOH на титрование, мл; значения Kсв и Kсл - см. выше.
Примечание. При наличии в воде сероводорода (H2S + HS-) и необходимости точного определения [CO2]об нужно специальным анализом определить общее содержание в воде соединений сероводорода (при pH менее 8,3 сумму H2S + HS-) и, учитывая величину pH воды, рассчитать концентрацию свободного сероводорода, мг-экв/л. Концентрацию свободной двуокиси углерода в мг/л в этом случае вычисляют по формуле
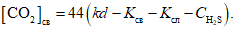
Определение концентраций свободной
и агрессивной двуокиси углерода по номограммам
7.8. Рассматриваемый метод можно применять в тех случаях, когда в воде помимо угольной кислоты и ее солей другие слабые кислоты и их соли содержатся в незначительных количествах.
Для определения содержания свободной двуокиси углерода [CO2]св и агрессивной двуокиси углерода [CO2]агр необходимы следующие данные:
а) температура воды - t, °C;
б) общее содержание солей - P, мг/л;
в) общая щелочность M; определяется титрованием соляной или серной кислотой в присутствии индикатора метилового оранжевого (см. стр. 41), мг-экв/л;
г) pH воды.
Содержание в воде свободной двуокиси углерода определяют по номограмме (рис. 4) в следующей последовательности:
1) соединяют линейкой на шкалах 1 и 3 деления, соответствующие значениям t и P; отмечают точку пересечения линейки с немой шкалой 2;
2) соединяют эту отметку с делением, отвечающим щелочности воды на шкале 4; отмечают точку пересечения прямой со шкалой 3, которая в этом случае используется в качестве немой;
3) полученную отметку на шкале 3 соединяют с соответствующим делением на шкале 5 (pH) и на продолжении прямой в месте ее пересечения со шкалой 6 находят искомую концентрацию свободной двуокиси углерода - [CO2]св.

свободной двуокиси углерода
Для определения содержания в воде агрессивной двуокиси углерода служат номограммы, приведенные на рис. 5 и 6.


агрессивной двуокиси углерода
Сначала по данным химического анализа воды на рис. 5 находят значение вспомогательной величины A. Для этого соединяют значения P и t и отмечают на немой шкале 4 точку, которую затем соединяют с величиной [Ca2+]. На пересечении прямой со шкалой 3 находят значение искомой величины A.
Зная величину A, концентрацию агрессивной двуокиси углерода находят по номограмме, приведенной на рис. 6.
На поле этой номограммы находят точку, соответствующую ранее определенным значениям [CO2]св и общей щелочности воды (M, мг-экв/л).
Если эта точка лежит выше кривой, отвечающей найденной величине A, то вода содержит агрессивную двуокись углерода, если ниже - агрессивной двуокиси углерода в воде нет.
Чтобы найти концентрацию агрессивной двуокиси углерода в первом приближении, нужно провести через эту точку с координатами M и [CO2]св прямую параллельно наклонным прямым, опускающимся слева направо, до пересечения с кривой, отвечающей величине A.
Ордината этой точки отвечает концентрации равновесной углекислоты [CO2]равн, а разность ординат [CO2]св и [CO2]равн дает концентрацию агрессивной углекислоты

 означает, что концентрация CO2 определена в первом приближении. При следующем уточнении ее обозначают
означает, что концентрация CO2 определена в первом приближении. При следующем уточнении ее обозначают  .
.Для уточнения полученного результата нужно подсчитать количество кальция, которое перейдет в раствор в результате растворения CaCO3 найденным количеством  по формуле
по формуле
по формуле
где [Ca2+]0 - содержание кальция в исследуемой воде согласно анализу, мг/л.
После этого снова определяют по номограмме (рис. 5) уточненную величину A, используя величину [Ca2+]' и прежние значения P и t. По уточненной величине A и прежним значениям [CO2]св и M по номограмме (рис. 6) находят указанным выше способом уточненные значения [CO2]равн и  .
.
.Обычно двух последовательных определений достаточно, чтобы определить [CO2]агр с нужной для практики степенью точности. При желании повысить точность определения можно выполнить его в третий раз.
Мешающие влияния и их устранение
7.9. Титрованию мешают окраска и мутность воды. В этом случае предпочтительно производить электрометрическое титрование. Если присутствует в воде активный хлор, происходит обесцвечивание индикатора. Для устранения влияния активного хлора к воде добавляют тиосульфат натрия в количестве, эквивалентном содержанию активного хлора.
При щелочности воды более 4 - 5 мг-экв/л в процессе титрования по фенолфталеину наблюдается часто помутнение раствора вследствие связывания едким натром равновесной углекислоты  , распада гидрокарбонат-ионов
, распада гидрокарбонат-ионов  и образования осадка CaCO3.
и образования осадка CaCO3.
, распада гидрокарбонат-ионов и образования осадка CaCO3.Распад гидрокарбонат-ионов приводит к тому, что помимо имеющейся в исследуемой воде свободной двуокиси углерода титруется двуокись углерода, выделяющаяся при распаде гидрокарбонатов, т.е. результат оказывается завышенным. Для устранения этого рекомендуется пробу разбавлять прокипяченной дистиллированной водой, чтобы щелочность пробы не превышала 3 - 4 мг-экв/л, либо определять свободную двуокись углерода, пользуясь номограммой.
Наличие в исследуемой воде слабых оснований, образующих осадок в результате гидролиза в процессе титрования, приводит к дополнительному расходу NaOH и нечеткому переходу окраски индикатора. В этом случае рекомендуется добавлять перед титрованием к пробе 0,8 - 1,2 мл раствора сегнетовой соли.
При необходимости получения особо точных результатов или при расходе на определение менее 0,5 мл раствора 0,1 н. NaOH рекомендуется проводить титрование 0,02 н. раствором NaOH.
Общие положения
8.1. Под общей щелочностью воды понимают сумму содержащихся в ней анионов гидрокарбонатных, карбонатных, гидратных и других слабых кислот, реагирующих с соляной или серной кислотой с образованием хлоридов или сульфатов.
Щелочность природных вод зависит в основном от содержания солей угольной кислоты. При цветности воды более 40° и необходимости точного определения концентрации гидрокарбонатных и карбонатных ионов следует отдельно учитывать величину гуматной щелочности (см. ниже).
Определение щелочности желательно производить непосредственно после отбора пробы воды, но не позднее чем через 24 ч, причем воду нужно хранить в сосуде, заполненном до пробки.
Определение общей щелочности
8.2. Общую щелочность определяют титрованием пробы исследуемой воды раствором соляной или серной кислоты до перехода окраски индикатора метилового оранжевого. Титрование можно проводить без индикатора, контролируя снижение pH в процессе титрования; заканчивается титрование при pH = 4,5.
При титровании имеют место следующие реакции с гидроксильными, карбонатными и гидрокарбонатными ионами:



Эти реакции завершаются при pH = 4,5; приблизительно этой же величине pH соответствует переход окраски индикатора метилового оранжевого от желтой к розовой.
При необходимости определения щелочности с точностью более 0,1 мг-экв/л титрование следует вести с выдуванием образующейся свободной двуокиси углерода воздухом.
Аппаратура
8.3. Бюретка для титрованного раствора кислоты.
pH-метр со стеклянным и каломельным электродами.
Устройство для продувания воды в колбе сжатым воздухом.
Реактивы
8.4. Соляная или серная кислота, 0,1 н. раствор; готовят из фиксанала или 8,25 мл концентрированной HCl пл. 1,19 ч.д.а. (или 2,8 мл H2SO4 пл. 1,84), доводят дистиллированной водой до 1 л.
Поправочный коэффициент 0,1 н. раствора кислоты определяют следующим образом: 1,9071 г буры, дважды перекристаллизованной и высушенной в течение 2 - 3 дней на воздухе между листами фильтровальной бумаги, растворяют в 100 мл дистиллированной воды в мерной колбе и доводят до метки. Берут калиброванной пипеткой точно 25 мл раствора буры, добавляют 3 капли раствора индикатора метилового оранжевого и титруют 0,1 н. раствором кислоты до перехода окраски.
Поправочный коэффициент определяют по формуле

где n - объем 0,1 н. раствора кислоты, израсходованной на титрование 25 мл раствора буры. Поправочный коэффициент вычисляют с точностью до 0,01.
Метиловый оранжевый (индикатор) - 0,1%-ный раствор (см. стр. 36).
Фенолфталеин (индикатор) - 0,5%-ный спиртовой раствор (см. стр. 36).
Едкий натр, 0,1 н. раствор; готовят из фиксанала либо растворением 4 г твердого едкого натра ч.д.а. в 1 л дистиллированной прокипяченной воды. Поправочный коэффициент к нормальности 0,1 н. раствора NaOH устанавливают по титрованному раствору 0,1 н. HCl (или 0,1 н. H2SO4):

где k1 - поправочный коэффициент 0,1 н. раствора NaOH;
k - поправочный коэффициент 0,1 н. раствора HCl (или 0,1 н. раствора H2SO4);
b - объем 0,1 н. раствора NaOH, взятый для определения поправочного коэффициента, мл;
a - объем 0,1 н. раствора HCl (или H2SO4), пошедший на титрование 0,1 н. раствора NaOH, мл.
Ход определения
8.5. К 100 мл исследуемой воды, отмеренным пипеткой в коническую колбу, добавляют 3 капли раствора метилового оранжевого и титруют 0,1 н. раствором кислоты до перехода окраски индикатора из желтой в золотисто-розовую. Можно проводить титрование без индикатора, контролируя величину pH титруемого раствора; титрование заканчивается при pH = 4,5.
При необходимости определения щелочности с точностью не менее 0,1 мг-экв/л после перехода окраски индикатора воду в колбе продувают воздухом и, в случае возврата окраски индикатора к первоначальной желтой, дотитровывают кислотой до перехода окраски в золотисто-розовую.
Расчет. Общую щелочность (M) в мг-экв/л вычисляют по формуле
M = ka1,
где k - поправочный коэффициент для приведения концентрации раствора соляной или серной кислоты к точно 0,1 н.;
a1 - расход 0,1 н. раствора кислоты на титрование, мл.
Определение компонентов общей щелочности
8.6. Основным компонентом щелочности природных вод при pH менее 8,3 являются анионы  (гидрокарбонатная, бикарбонатная щелочность), а при цветности более 40° также анионы гуминовой и фульвокислот (так называемая "гуматная" щелочность; при цветности 40° гуматная щелочность равна приблизительно 0,15 - 0,2 мг-экв/л).
(гидрокарбонатная, бикарбонатная щелочность), а при цветности более 40° также анионы гуминовой и фульвокислот (так называемая "гуматная" щелочность; при цветности 40° гуматная щелочность равна приблизительно 0,15 - 0,2 мг-экв/л).
При pH воды более 8,3 компонентами общей щелочности могут быть, в зависимости от величины pH, анионы  ,
,  , OH-, а при цветности более 40° при точных расчетах следует учитывать также гуматную щелочность.
, OH-, а при цветности более 40° при точных расчетах следует учитывать также гуматную щелочность.
Определение "гуматной" щелочности
8.7. Метод основан на свойстве гуматов при взаимодействии с сильной кислотой образовывать гуминовые кислоты, которые оттитровывают едким натром.
Ход определения
8.8. После определения общей щелочности по метиловому оранжевому добавляют к уже оттитрованной пробе 1 мл 0,1 н. раствора HCl (или 0,1 н. раствора H2SO4) и кипятят содержимое колбы 2 - 3 мин.
Затем быстро охлаждают колбу струей воды, добавляют 2 - 3 капли раствора фенолфталеина и титруют 0,1 н. раствором NaOH.
Записывают количество миллилитров 0,1 н. раствора NaOH, пошедшее на титрование от перехода окраски метилового оранжевого до перехода окраски фенолфталеина (m, мл).
Расчет. Гуматную щелочность воды (Г) в мг-экв/л вычисляют по формуле
Г = k1m,
где k1 - поправочный коэффициент для приведения концентрации раствора NaOH к точно 0,1 н.
Определение гидрокарбонатной, карбонатной
и гидратной щелочностей
8.9. Гидрокарбонатная, карбонатная и гидратная щелочности могут быть определены двумя методами: а) по общей щелочности, определенной с индикатором метиловым оранжевым (M), и щелочности, определенной с индикатором фенолфталеином (Ф); б) по номограммам (рис. 7 - 9).
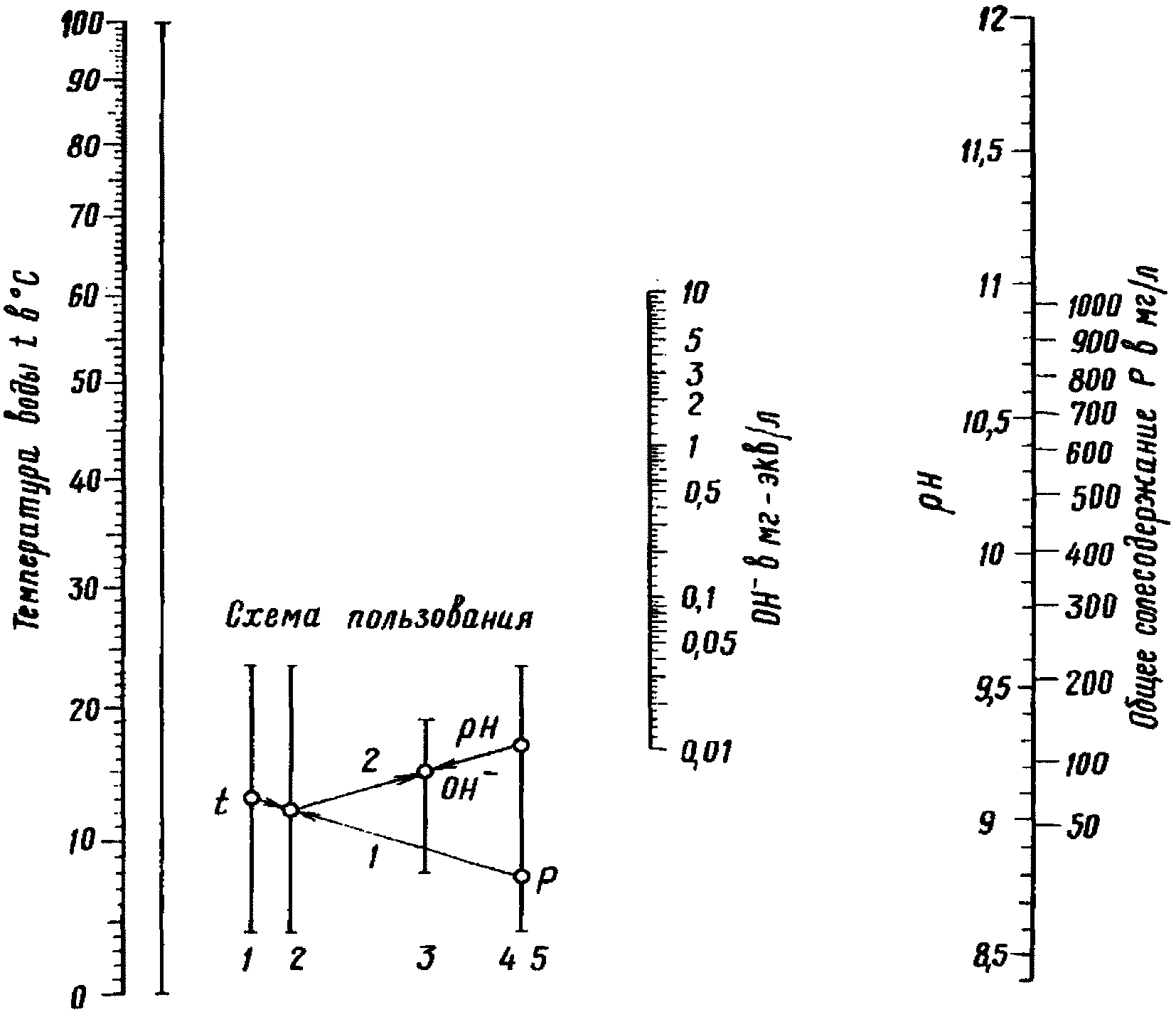

вспомогательной величины S

и карбонатной щелочностей воды
Определение гидрокарбонатной, карбонатной и гидратной
щелочностей по общей щелочности и щелочности,
найденной по фенолфталеину
8.10. Метод основан на том, что для вод с pH более 8,3 при титровании пробы исследуемой воды кислотой в присутствии индикатора фенолфталеина, дающего переход окраски от красного к бесцветному, протекают реакции между сильной кислотой, гидроксильными и карбонатными ионами, а в интервале pH от 8,3 до 4,5 (от перехода окраски фенолфталеина до перехода окраски метилового оранжевого) - с гидрокарбонатными ионами.
Ход определения щелочности по фенолфталеину
8.11. К 100 мл исследуемой воды, отмеренным пипеткой, в коническую колбу добавляют 5 - 10 капель раствора фенолфталеина и титруют 0,1 н. раствором кислоты (соляной или серной) до перехода окраски из красной в бесцветную.
Титрование можно проводить также без индикатора, заканчивая его, когда величина pH пробы воды снизится до 8,3.
Расчет. Щелочность воды по фенолфталеину (Ф) в мг-экв/л вычисляют по формуле
Ф = kn,
где k - поправочный коэффициент для приведения концентрации раствора кислоты (соляной или серной) к точно 0,1 н.;
n - расход 0,1 н. раствора кислоты, пошедшей на титрование с фенолфталеином, мл.
Определение гидратной, карбонатной и гидрокарбонатной щелочности производят либо приближенно по формулам табл. 6, либо более точно по номограммам (рис. 7 - 9).
Таблица 6
и гидрокарбонатной щелочностей воды
Соотношение между M1 и Ф | Вода содержит | Формула для расчета компонентов общей щелочности | ||
гидрокарбонат-ионы | карбонат-ионы | гидроксильные ионы OH- | ||
Ф = 0 | Гидрокарбонаты | M1 | 0 | 0 |
2Ф < M1 | Гидрокарбонаты и карбонаты | M1 - 2Ф | 2Ф | 0 |
2Ф = M1 | Карбонаты | 0 | 2Ф | 0 |
2Ф > M1 | Карбонаты и гидраты | 0 | 2(M1 - Ф) | 2Ф - M1 |
Ф = M1 | Гидраты | 0 | 0 | M1 |
При цветности воды более 40° и необходимости получения точных данных о компонентах общей щелочности сумму гидратной, карбонатной и бикарбонатной щелочностей (M1) в мг-экв/л, используемой в этих расчетах, определяют по формуле
M1 = M - Г.
Для приближенных расчетов можно считать M1 ~= M.
Определение гидратной, карбонатной и гидрокарбонатной
щелочностей по номограммам
8.12. Исходным для определения являются следующие данные анализов исследуемой воды: а) сумма OH-,  и
и  [M1 по формуле на стр. 45); б) общее содержание в воде солей (приближенно сухой остаток); в) величина pH; 4) температура воды.
[M1 по формуле на стр. 45); б) общее содержание в воде солей (приближенно сухой остаток); в) величина pH; 4) температура воды.
Определение гидратной щелочности воды (OH- в мг-экв/л) производят по номограмме, приведенной на рис. 7. Соединяют линейкой деление на шкале 1 (температура воды) с делением на шкале 5 (общее солесодержание). Отмечают точку пересечения прямой с немой шкалой 2, затем эту точку соединяют прямой с величиной pH воды на шкале 4. На пересечении прямой со шкалой 3 находят искомую величину гидратной щелочности воды.
Карбонатную и гидрокарбонатную щелочность воды определяют по номограммам, приведенным на рис. 8 - 9. Для этого предварительно определяют сумму карбонатной и гидрокарбонатной щелочности в мг-экв/л по формуле
N = M1 - OH-.
По номограмме (рис. 8) определяют вспомогательную величину S. Для этого соединяют линейкой деления на шкалах 1 и 5, соответствующие содержанию солей и температуре воды, и отмечают точку пересечения с немой шкалой. Эту точку соединяют с величиной pH на шкале 2 и на пересечении прямой со шкалой 3 находят величину S. Затем на рис. 9 - соединяют линейкой значение S на шкале 1 со значением N на шкале 3 и на пересечении прямой со шкалой 2 находят искомую величину (гидрокарбонатная щелочность). Далее соединяют значение на шкале 3 с найденной ранее величиной S на шкале 5. Пересечение прямой со шкалой 4 дает искомую величину  (карбонатная щелочность).
(карбонатная щелочность).
Мешающие влияния и их устранение
8.13. При общей щелочности воды менее 0,4 мг-экв/л титрование 0,1 растворами HCl или H2SO4 дает недостаточно точные результаты. В этом случае целесообразно производить титрование 0,05 н. раствором кислоты.
При наличии в исследуемой воде взвешенных веществ перед титрованием пробу следует профильтровать через беззольный бумажный или стеклянный фильтр. Удаление взвеси необходимо в связи с тем, что в ее составе могут быть карбонаты (CaCO3, MgCO3) и другие вещества, на растворение которых расходуется кислота.
При интенсивной окраске воды, мешающей визуальному определению перехода окраски индикаторов, рекомендуется производить титрование без индикаторов.
При невозможности электрометрического титрования воду для устранения окраски фильтруют через слой зернистого активного угля, загруженного в стеклянную трубку.
Наличие в воде свободного хлора приводит к обесцвечиванию индикатора. Для устранения этого к воде добавляют раствор тиосульфата натрия в количестве, эквивалентном содержанию свободного хлора, либо воду фильтруют через слой зернистого активного угля, загруженного в стеклянную трубку.
Свободная двуокись углерода, содержащаяся в исследуемой воде, так же как и двуокись углерода, выделяющаяся в процессе титрования, является источником погрешности при определении перехода окраски индикатора, а также при определении конца титрования по pH-метру. Для повышения точности определения общей щелочности воды в необходимых случаях продувают воду во время титрования воздухом, как это указано выше (стр. 42).
При наличии в воде кроме гуматных также других анионов слабых кислот (силикатных, сульфидных, фосфатных и др.) они титруются сильной кислотой совместно с гидроксильными и карбонатными ионами.
В случае необходимости последующего расчета концентраций ионов OH-,  и
и  по табл. 6 или номограммам (рис. 7 - 9) из определенной титрованием величины общей щелочности воды нужно вычесть сумму концентраций (в мг-экв/л) гуматов и других слабых кислот, концентрации которых определяются по соответствующим методикам (см. определение кремнекислоты, сульфидов, фосфатов).
по табл. 6 или номограммам (рис. 7 - 9) из определенной титрованием величины общей щелочности воды нужно вычесть сумму концентраций (в мг-экв/л) гуматов и других слабых кислот, концентрации которых определяются по соответствующим методикам (см. определение кремнекислоты, сульфидов, фосфатов).
Общие положения
9.1. Небольшая часть молекул воды диссоциирована на водородные и гидроксильные ионы. В химически чистой воде молярные концентрации упомянутых ионов равны друг другу, составляя при 25 °C 10-7 мол/л. Таким образом, величина произведения обеих концентраций равна 10-14. Это произведение сохраняет постоянную величину и в присутствии веществ, при диссоциации которых освобождаются водородные и гидроксильные ионы. Поэтому вполне достаточно определить концентрацию одного из них. Практически определяют концентрацию водородных ионов.
Поскольку концентрация водородных ионов может достигать весьма различной величины в пределах различных порядков, принято выражать ее величиной pH, представляющей отрицательный десятичный логарифм:
[H+] = 10-pH; pH = -lg[H+].
Определение концентрации водородных ионов осуществляется в интервале от 1 до 10-14 мол/л, что соответствует величине pH от 0 до 14. Величина pH = 7 отвечает нейтральному состоянию раствора, понижение этой величины - кислотному, а повышение - щелочному.
Величина pH является важным показателем кислотности или щелочности воды и показывает содержание веществ, которые вызывают соответствующую кислотность или щелочность. Она служит вспомогательной величиной при различных аналитических расчетах.
В большинстве природных вод концентрация водородных ионов обусловлена лишь отношением между концентрациями свободной двуокиси углерода и гидрокарбонат-ионов. В этих случаях значение pH колеблется от 4,5 до 8,3. На величину pH может оказать влияние повышенное содержание гуминовых веществ, основных карбонатов и гидроокисей, образующихся вследствие поглощения CO2 в процессе фотосинтеза, а в отдельных случаях - также и повышенное содержание солей, подверженных гидролизу, и пр. В загрязненных поверхностных водах, кроме этого, могут содержаться и другие вещества, в том числе сильные кислоты и основания.
Величину pH определяют электрометрическим методом, измеряя потенциал, возникающий на соответствующем измерительном электроде.
Учитывая, что pH часто является результирующей величиной равновесия веществ переменного состава, рекомендуется производить определение немедленно после отбора пробы. Если это не выполнимо, то рекомендуется доставлять пробу к месту анализа в особой бутылке, наполненной по способу, описанному при определении кислотности (стр. 15), предохраняя ее от нагревания и обрабатывая в кратчайший срок.
Результаты определений выражаются в pH и лишь в исключительных случаях (см. расчетное определение, стр. 52) - в миллиграмм-эквивалентах водородных или гидроксильных ионов в 1 л.
Электрометрическое определение pH
9.2. Электрометрическое определение pH со стеклянным электродом основано на том, что изменение значения pH на единицу в определенной области pH вызывает изменение потенциала электрода на 58,1 мВ при 20 °C. Пределы линейной зависимости потенциала электрода от pH зависят от состава стекла электрода.
Мешающие влияния
9.3. Результат определения зависит от температуры пробы. Влияние температуры компенсируется специальным устройством, вмонтированным в прибор. Пробу воды подогревают или охлаждают, устанавливая ее температуру в 20 °C. Если температура пробы незначительно отличается от 20 °C и ее не приводят к 20 °C, то при записи результатов определения нужно указать температуру опыта.
Электрометрическому измерению не мешают окраска, мутность, взвесь, свободный хлор, присутствие окисляющих или восстанавливающих веществ или же повышенное содержание солей в пробе.
Некоторые помехи возникают при повышенном содержании солей натрия и при pH больше 10. В таких случаях необходимо пользоваться специальным электродом или же вводить поправки, указанные в инструкции, приложенной к электроду.
Точность электрометрического определения снижается при пользовании загрязненными электродами. Для исследования сильно загрязненных проб следует иметь отдельный электрод, применяемый только для этой цели. Если возникает необходимость обезжирить электрод, то пользуются куском тонкой материи, смоченной эфиром или раствором синтетического моющего вещества. После этого необходимо несколько раз промыть электрод дистиллированной водой, вытирая его каждый раз для удаления обезжиривающего вещества. При необходимости действие электрода восстанавливают погружением его на 2 ч в 2%-ный раствор соляной кислоты с тщательной последующей промывкой дистиллированной водой. В нерабочее время электрод следует хранить в дистиллированной воде.
Аппаратура
9.4. Лабораторный pH-метр (потенциометр) со стеклянным электродом измерения и каломельным электродом сравнения.
Реактивы
9.5. Буферный раствор: pH = 1,68 (20 °C); растворяют 12,710 г биоксалата калия KH3(C2O4)2·2H2O ч.д.а. в свежепрокипяченной и охлажденной дистиллированной воде и доводят при 20 °C объем до 1 л.
Буферный раствор: pH = 4,00 (20 °C); растворяют 10,211 г бифталата калия C6H4·COOK·COOH ч.д.а., высушенного при 110 °C, в свежепрокипяченной и охлажденной дистиллированной воде и доводят объем при 20 °C до 1 л.
Буферный раствор: pH = 6,98 (20 °C); растворяют одновременно в свежепрокипяченной и охлажденной дистиллированной воде 1,361 г KH2PO4 ч.д.а. и 1,420 г Na2HPO4 ч.д.а., высушенных при температуре 110 - 130 °C, и доводят объем при 20 °C до 1 л.
Буферный раствор: pH = 9,22 (20 °C); растворяют в свежепрокипяченной и охлажденной дистиллированной воде 3,814 г Na2B4O7·10H2O ч.д.а., сохраняемого продолжительное время в эксикаторе над бромидом натрия, и доводят объем до 1 л при 20 °C.
Буферные растворы: pH = 9,97; 11,08; 12,30; растворяют в свежепрокипяченной и охлажденной дистиллированной воде 19,108 г Na2B4O7·10H2O ч.д.а., сохраняемого продолжительное время в эксикаторе над бромидом натрия, и доводят объем до 1 л при 20 °C. Затем следует приготовить приблизительно 0,1 н. раствор едкого натра (приготовление см. стр. 35); раствор доводят точно до 0,1 н. исходя из результатов титрования 20 мл раствора NaOH точным 0,1 н. раствором соляной кислоты (приготовление см. стр. 42) с индикатором метиловым оранжевым. Пользуясь точными бюретками, составляют следующие смеси: 4,0 - 5,0 - 6,0 мл 0,1 н. раствора NaOH + 6,0 - 5,0 - 4,0 мл раствора Na2B4O7. Полученные в результате смешения растворы имеют при 18 °C значения pH соответственно 9,97; 11,08; 12,30.
Калибровочная кривая
9.6. Если прибор имеет шкалу только в милливольтах, то необходимо произвести калибровку измерительных электродов с помощью буферных растворов с известным значением pH. Строят график зависимости найденных значений потенциалов от величины pH использованных буферных растворов.
Ход определения
9.7. Перед началом измерения электрод промывают дистиллированной водой, затем исследуемой водой и лишь потом погружают в анализируемую пробу. Пробу следует предварительно тщательно перемешать, чтобы ее состав непосредственно у поверхности электрода соответствовал общему ее составу. Температуру пробы перед определением не устанавливают. Одновременно с электродами в пробу погружают промытый в пробе термометр для определения ее температуры во время измерения и внесения необходимых поправок. Измеряемую величину потенциала стеклянного электрода отсчитывают в милливольтах или прямо в единицах pH. Метод измерений зависит от типа применяемого прибора и указывается в приложенных к нему инструкциях.
Округление результатов. Полученные результаты принято округлять до 0,05 - 0,1 pH в зависимости от типа применяемого прибора, указав при записи, что измерения проводились электрометрическим методом.
Расчет концентрации водородных и гидроксильных ионов
9.8. Концентрацию водородных и гидроксильных ионов можно рассчитать на основании результатов определения значения pH, кроме того, для гидроксильных ионов можно пользоваться результатами определения общей (m) и свободной (p) щелочности.
Выполнение такого рода расчетов возможно во всех случаях, но результаты их приводят лишь тогда, когда получаются величины, имеющие практическое значение. Если эти величины достигают порядка 0,01 миллиграмм-эквивалента на 1 л, то их можно использовать при составлении баланса анионов и катионов. Концентрации водородных ионов, выраженные в миллиграмм-эквивалентах, имеют значение, начиная от pH = 5 и ниже, а концентрации гидроксильных ионов - начиная от pH = 9 и выше. Расчет содержания гидроксильных ионов по щелочности можно считать ориентировочным (см. стр. 46). Если свободная щелочность не превышает половины общей щелочности, то принято считать концентрацию гидроксильных ионов столь малой, что ею можно пренебречь.
Расчет. Для расчета пользуются формулами:
водородные ионы
[H+] = 103 - pH мг-экв/л;
гидроксильные ионы
[OH-] = 103 - pKw + pH мг-экв/л,
где pKw - отрицательный логарифм константы, называемой "ионное произведение воды" (численные значения указаны в химических справочниках, например см. Ю.Ю. Лурье "Справочник по аналитической химии". "Химия", 1971). При 25 °C pKw = 14.00.
Округление результатов
Интервал, мг-экв/л | 0,01 - 2,00 | 2,00 - 5,00 | 5,00 - 10,00 | 10,00 - 20,00 |
Округление, мг-экв/л | 0,01 | 0,02 | 0,05 | 0,1 |
Общие положения
10.1. Общая жесткость воды показывает концентрацию в ней катионов двухвалентных щелочноземельных металлов, прежде всего кальция и магния. Эти элементы в природных условиях попадают в воду вследствие воздействия углекислого газа на карбонатные минералы или за счет продуктов микробиального распада, происходящего в увлажненных слоях почвы. Естественное содержание солей, вызывающих жесткость воды, может измениться в результате соответствующей обработки, а в водотоках - сбросом некоторых видов сточных вод.
Количество кальция и магния, эквивалентное количеству карбонатов и бикарбонатов, называется карбонатной жесткостью. Некарбонатная жесткость определяется как разность между общей и карбонатной жесткостью и показывает количество катионов щелочноземельных металлов, соответствующих анионам минеральных кислот: хлоридам, сульфатам, нитратам и др.
Общая жесткость определяется в питьевых, подземных и поверхностных водах. Для определения общей жесткости вод всех видов ниже приведен комплексонометрический метод.
Для определения карбонатной и некарбонатной жесткости пользуются расчетом.
Результаты определения жесткости выражаются в мг-экв/л.
Пробы для определения жесткости не консервируют. Сильнокислые или сильнощелочные пробы перед исследованием нейтрализуют.
Комплексонометрический метод определения
общей жесткости воды
10.2. Способность аминополикарбонатных кислот, таких, как иминотриуксусная, иминодиуксусная, этилендиаминтетрауксусная и др., образовывать комплексные анионы с ионами щелочноземельных металлов (и некоторыми другими двухвалентными катионами) позволила применить эти кислоты для определения жесткости воды. Практически было предложено применять для этой цели двузамещенную натриевую соль этилендиаминтетрауксусной кислоты, образующую в водной среде при pH = 10 прочные комплексные соединения - сначала с ионами кальция, затем с ионами магния. Для определения жесткости применяют титрование раствором комплексона III (трилона Б), который имеет следующее строение:
 .
.Реакции комплексообразования, происходящие при титровании воды, содержащей ионы кальция или магния, могут быть выражены уравнением
 .
.Как видно из уравнения, ионы кальция и магния полностью связываются в комплексные соединения, т.е. как бы удаляются из раствора.
Переход обоих ионов в комплексные соединения сопровождается изменением окраски индикатора эриохромчерного T. Комплекс эриохромчерного T с ионами магния имеет интенсивный вишнево-красный цвет, свободная форма индикатора имеет синюю окраску. Определение будет правильным, если титруемый раствор имеет pH = 10 +/- 0,1 и достаточное количество ионов магния. При  значениях pH может выделяться в осадок Mg(OH)2, а при более низких величинах pH магний недостаточно прочно связан с красителем.
значениях pH может выделяться в осадок Mg(OH)2, а при более низких величинах pH магний недостаточно прочно связан с красителем.
значениях pH может выделяться в осадок Mg(OH)2, а при более низких величинах pH магний недостаточно прочно связан с красителем.Точность определения при титровании 100 мл пробы составляет 0,05 мг-экв/л, что является также и минимально определяемой концентрацией.
10.3. Предельные концентрации ионов, не искажающие результатов определения жесткости комплексонометрическим методом, представлены в табл. 7. Из таблицы видно, что комплексонометрическим методом можно определять жесткость в присутствии больших количеств хлоридов, сульфатов, бихроматов и многих других ионов. Ионы меди, марганца и цинка мешают определению, но их вредное влияние можно устранить, удалив медь и цинк в виде сульфидов; влияние марганца устраняют гидроксиламином.
Таблица 7
при комплексонометрическом определении жесткости
Ионы | Предельно допустимые концентрации в мг·л | Ионы | Предельно допустимые концентрации в мг/л |
Бихроматы | 1000 | Алюминий Al3+ | 10 |
Карбонаты | 1000 | Железо Fe2+ | 10 |
Нитраты | 600 | Железо Fe3+ | 10 |
Нитриты | 500 | Кадмий Cd2+ | Титруется, как соли жесткости |
Силикаты | 100 | Кобальт Co2+ | 0,1 |
Сульфаты | 10 000 | Марганец Mn2+ | Титруется, как соли жесткости |
Сульфиты | 500 | Медь Cu2+ | 0,5 |
Хлориды Cl- | 10 000 | Никель Ni2+ | 0,1 |
Цинк Zn2+ | Титруется, как соли жесткости | ||
Сульфат-целлюлозный щелок | 100 |
Если в воде присутствуют значительные количества бихроматов, красная окраска раствора в конечной точке титрования переходит не в синий, а в ярко-зеленый цвет.
Поскольку дистиллированная вода может быть загрязнена ионами некоторых металлов (например, меди, цинка), препятствующих определению, для приготовления растворов, применяемых в данном методе, следует использовать дистиллированную воду, не содержащую указанных ионов. Для этой цели в случае надобности дистиллированную воду подвергают H-катионированию или повторной перегонке из стеклянной или другой посуды, не обогащающей дистиллят ионами указанных металлов.
Если в сосуде с пробой в промежуток времени между ее взятием и обработкой выделится углекислый кальций, то пробу надо обработать способом, описанным на стр. 115.
Неясный переход окраски индикатора происходит вследствие присутствия металлов, комплексы которых с примененным индикатором более прочны, чем с комплексоном III. Определению жесткости мешают присутствие железа (10 мг/л), кобальта (0,1 мг/л), никеля (0,1 мг/л) и меди (0,5 мг/л). Другие катионы, как, например, свинец, кадмий, марганец, цинк, барий и стронций, титруются вместе с кальцием и магнием и повышают этим расход титрованного раствора комплексона III. Для устранения мешающих влияний при титровании и для связывания некоторых катионов, вызывающих повышенный расход раствора, можно применить цианид калия, гидроксиламин солянокислый или сульфид натрия, которые прибавляют к титруемому раствору.
Цианид калия прибавляют в виде сухой соли в количестве от 0,25 до 0,5 г на 100 мл пробы перед прибавлением индикатора. В этом случае исключаются мешающие при титровании ионы: кобальт, никель и алюминий - до 200 мг/л, медь и железо - до 300 мг/л. Одновременно титруются барий, кадмий, свинец, марганец, стронций и цинк.
Раствор гидроксиламина солянокислого приготовляется растворением 4,5 г этого вещества в 100 мл этилового или изопропилового спирта. Прибавляют от 5 до 10 капель его к 100 мл пробы после приведения pH к нужной величине и то только тогда, когда гидроксиламин солянокислый не является составной частью раствора индикатора. В этом случае при титровании исключается мешающее влияние меди до 0,3 мг/л, марганца - до 1 мг/л, железа и алюминия - до 20 мг/л. Одновременно титруются барий, кадмий, свинец, стронций и цинк. Мешающее влияние кобальта и никеля этим способом не устраняется.
Раствор сульфида натрия приготовляют растворением 5 г Na2S·9H2O или 3,7 г Na2S·5H2O в 100 мл дистиллированной воды. До прибавления индикатора к 100 мл титруемой пробы прибавляют 2 мл раствора сульфида натрия. Тогда при титровании не мешают цинк до 200 мг/л, алюминий, кадмий и свинец - до 20 мг/л, железо - до 5 мг/л, марганец - до 1 мг/л, кобальт и никель - до 1,3 мг/л. Одновременно титруются барий и стронций.
Взвешенные и коллоидные вещества, находясь в большом количестве, препятствуют определению. Для обычных проб воды вполне достаточно фильтрования, для сильно загрязненных вод надо определенное количество фильтрованной пробы выпарить досуха, прокалить при температуре 600 °C, остаток растворить в 40 мл 1 н. раствора HCl при нагревании, после растворения и фильтрования нейтрализовать 3 н. раствором NH4OH до получения pH = 7 и долить дистиллированной водой до первоначального объема.
Сильнокислые или сильнощелочные пробы перед исследованием нейтрализуют.
Реактивы
10.4. Аммиак + хлорид аммония, буферный раствор; pH = 10; растворяют 26,8 г NH4Cl ч.д.а. в 100 мл дистиллированной воды и смешивают с 300 мл концентрированного раствора аммиака ч.д.а. К полученному раствору прибавляют 50 мл 0,05 М раствора сульфата магния (1,232 г MgSO4·7H2O ч.д.а. в 100 мл дистиллированной воды) и эквивалентное количество 0,05 М раствора комплексона III, которое определяется титрованием раствора сульфата магния способом, описанным на стр. 118.
Проверку величины pH раствора производят следующим образом:
а) к 100 мл дистиллированной воды прибавляют 5 мл буферного раствора и электрометрически измеряют величину pH, которая должна иметь значение 10 +/- 0,1. Если последняя оказалась меньше, титруют раствором аммиака, при более высоком значении pH - соляной кислотой до получения pH = 10. После пересчета расхода и прибавления соответствующего количества аммиака или кислоты к оставшемуся буферному раствору производят новую проверку;
б) к следующим 100 мл дистиллированной воды прибавляют точно отмеренные пипеткой 5 мл буферного раствора и 5 капель раствора индикатора эриохромчерного T. При этом должно появиться грязно-сине-фиолетовое окрашивание, которое после прибавления одной капли 0,05 М раствора комплексона III должно перейти в чисто-синее. Если расход комплексона III будет более высоким, необходимо на каждые 5 мл буферного раствора прибавить такое количество комплексона III, которое было израсходовано при проверочном испытании для достижения необходимого изменения окраски за вычетом 0,03 мл. Если после прибавления индикатора возникло синее окрашивание, то прибавляют 0,05 М раствор сульфата магния до возникновения сине-фиолетового окрашивания. После пересчета прибавляют соответствующее количество раствора сульфата магния к общему объему буферного раствора. После обеих обработок производят новую проверку.
Эриохромчерный T, раствор индикатора или сухая смесь:
а) раствор приготовляют смешением 0,5 г эриохромчерного T с 4,5 г гидроксиламина солянокислого и растворением в 100 мл 96%-ного этилового или изопропилового спирта. Раствор сохраняют в темноте, в тщательно закрытой склянке и выдерживают 6 недель;
б) смесь индикатора с хлоридом натрия приготовляют смешением и тщательным растиранием 0,5 г эриохромчерного T со 100 г хлорида натрия ч.д.а.
Комплексон III, 0,05 М раствор: 18,6 г комплексона III (двунатриевой соли этилендиаминтетрауксусной кислоты) ч.д.а. растворяют в дистиллированной воде и объем доводят до 1 л. Титр или поправку определяют по стандартному 0,025 М раствору хлорида кальция; 20 мл этого раствора в колбе для титрования разбавляют до 100 мл дистиллированной водой и обрабатывают соответственно ходу определения анализируемой пробы. Титр или поправку проверяют 1 раз в неделю.
Хлорид кальция, 0,025 М раствор: 2,5023 г свежеосажденного CaCO3 ч.д.а., высушенного при 105 °C, растворяют в конической колбе в требуемом количестве соляной кислоты (1:1). После полного растворения прибавляют около 200 мл дистиллированной воды, нагревают и кипятят около 5 мин. После охлаждения и прибавления нескольких капель метилового красного обрабатывают 3 н. раствором аммиака или соляной кислотой до перехода окраски индикатора. Объем раствора в мерной колбе доводят до 1 л.
Ход определения
10.5. К 100 мл пробы воды или к другому объему, доведенному до 100 мл, приливают 5 мл буферного раствора. Мешающее влияние щелочности устраняют предварительно добавлением эквивалентного количества 0,1 н. раствора соляной кислоты. После тщательного перемешивания прибавляют около 5 капель раствора индикатора или 0,1 - 0,2 г сухой смеси индикатора с NaCl. После перемешивания раствор титруют 0,05 М раствором комплексона III до перехода красной окраски в фиолетовую. Затем по каплям при тщательном перемешивании постепенно доводят титрование до синей окраски исследуемого раствора. Изменение окраски подтверждают прибавлением еще 1 капли (0,02 мл) титрованного раствора. При неясном переходе окраски определение повторяют после предварительной обработки пробы, описанной в разделе "Мешающие влияния". Титрование не должно продолжаться больше 5 мин.
Расчет. Общую жесткость (x) в мг-экв/л вычисляют по формуле
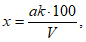
где a - количество израсходованного 0,05 М титрованного раствора комплексона III, мл;
k - поправка молярности раствора комплексона III;
V - количество взятой для титрования пробы, мл.
Округление результатов
Диапазон, мг-экв/л | 0,05 - 1,00 | 1,00 - 2,00 | 2,0 - 5,0 | 5,0 - 10,0 |
Округление, мг-экв/л | 0,02 | 0,05 | 0,1 | 0,2 |
Расчет карбонатной и некарбонатной жесткости воды
10.6. Для вычисления карбонатной жесткости используют результаты определения щелочности. У проб, которые содержат большое количество катионов щелочных металлов и у которых часть гидрокарбонатных и карбонатных ионов связана с этими металлами, величина, вычисленная по щелочности, не отвечает карбонатной жесткости. В этих случаях имеется в виду только общая жесткость.
При анализе вод, щелочность которых вызвана не только исключительно ионными формами угольной кислоты, а также и анионами других слабодиссоциированных кислот, в результате расчета будет ошибка, пропорциональная концентрации этих веществ.
Карбонатная жесткость (в мг-экв/л) равна m, где m - общая щелочность в мг-экв/л.
Некарбонатная жесткость (в мг-экв/л) равна разнице между общей жесткостью и карбонатной жесткостью.
Округление результатов
Результаты определения карбонатной и некарбонатной жесткости округляют так же, как и при комплексонометрическом определении.
Общие положения
11.1. В природных водах содержатся в тех или иных количествах органические вещества в коллоидном или истинно-растворенном состоянии.
Загрязнение воды этими веществами происходит за счет продуктов распада растительных и животных организмов, либо в результате сброса промышленных и бытовых сточных вод.
Предварительная обработка воды на водоочистных сооружениях только частично устраняет указанные загрязнения. Каждая вода содержит легко- и трудноокисляющиеся органические вещества и в зависимости от степени загрязнения эти вещества могут быть окислены сильными окислителями - перманганатом, бихроматом и др.
Количество кислорода, эквивалентное расходу окислителя, называется окисляемостью. По окисляемости можно приблизительно установить содержание органических веществ в воде.
Окисляемость выражают обычно в миллиграммах кислорода, пошедшего на окисление примесей, содержащихся в 1 л воды.
В зависимости от применяемого окислителя различают окисляемость перманганатную и бихроматную. Окисляемость воды перманганатом косвенно характеризует концентрацию легкоокисляющихся органических веществ в воде.
Окисляемость чистых подпочвенных и родниковых вод обычно не превышает 1 - 3 мг/л O2; в водах поверхностных источников окисляемость достигает 10 - 12 мг/л O2. Более высокая окисляемость, достигающая 25 - 30 мг/л O2, указывает на загрязнение воды.
В водах торфяных и болотистых, содержащих гуминовые вещества, окисляемость может быть еще выше.
Для определения перманганатной окисляемости питьевых, поверхностных и малозагрязненных вод применяют методы Кубеля (определение окисляемости в кислой среде) и Шульце-Паппа (в щелочной среде).
Для определения окисляемости поверхностных вод, загрязненных трудноокисляемыми веществами, рекомендуется бихроматный метод.
Пробы консервируют добавлением 2 мл разбавленной H2SO4, (1:2) на 100 мл пробы.
Пробы питьевых вод консервируют, если они не анализируются в течение 48 ч; пробы поверхностных вод консервируют, если их не предполагается анализировать в течение суток; пробы сильно загрязненных поверхностных вод консервируют в том случае, если их не будут анализировать в этот же день.
Перманганатный метод (метод Кубеля)
11.2. Метод основан на окислении веществ, присутствующих в пробе воды, 0,01 н. раствором перманганата калия в сернокислой среде при кипячении.
Без разбавления можно определять окисляемость до 10 мг кислорода в 1 л воды. Наивысшее допустимое разбавление проб - десятикратное. Это означает, что метод можно использовать только для проб, окисляемость которых ниже 100 мг кислорода в 1 л. Для проб с более высокой окисляемостью пользуются другим методом, например так называемым "четырехчасовым окислением" (см. стр. 62).
Окисление перманганатом калия в кислой среде протекает по уравнению

После кипячения раствора в течение установленного времени к нему прибавляют щавелевую кислоту, восстанавливающую избыток перманганата калия.

Избыток прибавленной щавелевой кислоты затем оттитровывают раствором перманганата калия.
Мешающие влияния
11.3. При определении окисляемости, дающей приблизительное представление о содержании в пробе окисляемых органических веществ, все же необходимо устранить мешающие влияния неорганических соединений, которые также могут быть окисленными при определении. К таким соединениям относятся хлориды, сульфиды, нитриты, двухвалентное железо. Когда концентрация хлоридов даже после разбавления пробы превышает 300 мг/л, для определения окисляемости применяется метод Шульце-Паппа. Двухвалентное железо, сероводород, сульфиды и нитриты следует определять отдельно, и результат, пересчитанный на окисляемость (мг O2), вычесть из найденной величины окисляемости пробы; при этом пользуются следующими соотношениями: 1 мг H2S соответствует 0,47 мг O2; 1 мг  - 0,35 мг O2 и 1 мг Fe2+ - 0,14 мг O2.
- 0,35 мг O2 и 1 мг Fe2+ - 0,14 мг O2.
Аппаратура
11.4. Конические колбы для кипячения, плоскодонные, емкостью от 250 до 300 мл, предназначенные только для определения окисляемости. Новые колбы должны быть обработаны горячим раствором перманганата.
Стеклянные шарики или обожженная пемза, также обработанные горячим раствором перманганата.
Реактивы
11.5. Серная кислота, разбавленный раствор: 1 объем 96%-ной ч.д.а. прибавляют при перемешивании к 2 объемам дистиллированной воды. К полученному раствору добавляют 0,01 н. раствор перманганата до слабо-розовой окраски и кипятят 30 мин, добавляя еще раствор перманганата, если розовая окраска исчезнет.
Щавелевая кислота, 0,1 н. основной раствор; готовят из фиксанала или 6,3030 г (COOH)2·2H2O ч.д.а.; растворяют в разбавленной (1:15) серной кислоте и серной кислотой того же разбавления доводят до 1 л при температуре 20 °C. Раствор сохраняют в темной бутыли. Устойчив около полугода.
Щавелевая кислота, 0,01 н. раствор: 100 мл 0,1 н. основного раствора щавелевой кислоты доводят до 1 л разбавленной (1:15) серной кислотой. Титрованный раствор можно также приготовить растворением 0,7303 г (COOH)2·2H2O ч.д.а. в разбавленной (1:15) серной кислоте и доведением той же кислотой (1:15) до 1 л.
Перманганат калия, приблизительно 0,1 н. запасной раствор: 3,2 г KMnO4 растворяют в 1 л дистиллированной воды, сохраняют в темной бутыли, изредка перемешивая. Раствор можно применять не раньше чем через 2 - 3 недели. При образовании осадка после отстаивания раствор осторожно сливают, пользуясь стеклянным сифоном, или отфильтровывают через стеклянную вату.
В случае необходимости отстаивание раствора заменяют кипячением в течение 1 ч. Колбу, в которой кипятят раствор, предохраняют от попадания в нее пыли. После охлаждения раствор отфильтровывают через чистую стеклянную вату или волокнистый асбест (предварительно прокаленный). Можно использовать фильтрующий тигель или воронку с пористой пластинкой. Фильтрат собирают в чистую бутыль из темного стекла, предварительно тщательно очищенную хромовой смесью.
Перманганат калия, 0,01 н. раствор: готовят разбавлением 0,1 н. раствора в 10 раз (по объему) дистиллированной водой.
Приготовленному раствору дают отстояться несколько дней и отделяют его от осадка. Можно приготовить 0,01 н. раствор, растворяя 0,32 г KMnO4 в 1 л воды.
Растворы перманганата 0,01 н. концентрации неустойчивы и не пригодны для длительного хранения. Поэтому периодически перед работой необходимо проверять титр этих растворов.
Хранят растворы перманганата калия в бутылях из темного стекла в защищенном от солнечного света месте.
При работе с растворами перманганата нельзя употреблять резиновые трубки и пробки.
Бюретки для титрования должны быть со стеклянными кранами.
Дистиллированная вода, дважды перегнанная, не содержащая окисляющихся веществ.
Ход определения
11.6. В коническую колбу для кипячения помещают несколько стеклянных шариков или кусочков пемзы, наливают 100 мл пробы или меньшее ее количество (в этом случае объем доводят до 100 мл дистиллированной водой), приливают 5 мл разбавленной серной кислоты и 10 мл 0,01 н. раствора перманганата.
В колбу вставляют маленькую воронку с отрезанной трубкой, нагревают жидкость до кипения (это время должно быть не более 5 мин) и с этого момента кипятят точно 10 мин.
После этого снимают колбу с огня, вливают в нее 10 мл раствора щавелевой кислоты и обесцвеченную жидкость титруют раствором перманганата калия до слабо-розового окрашивания. Температура смеси при титровании не должна падать ниже 80 °C.
Расход перманганата отсчитывают с точностью 0,05 мл. Если раствор при кипячении обесцветится или побуреет, определение нужно повторить с разбавленной пробой.
Определение следует повторить и тогда, когда перманганата расходуется более 60% добавленного количества, т.е. расход на титрование превышает 6 мл.
При титровании разбавленных проб не должно быть израсходовано меньше 20% добавленного количества перманганата, т.е. около 2 мл.
Для определения титра перманганата калия в ту же колбу после титрования к еще горячей жидкости приливают 10 мл 0,01 н. раствора щавелевой кислоты и снова оттитровывают перманганатом калия до такой же окраски, сохраняющейся в течение 1 мин.
Разделив 10 на число миллилитров раствора перманганата калия, израсходованного при втором титровании, получают поправку (k) для приведения концентрации раствора KMnO4 к точно 0,01 н. Если определение производится с разбавлением пробы, то параллельно с определением окисляемости анализируемой воды таким же образом определяют окисляемость дистиллированной воды, которая была применена для разбавления анализируемой воды.
Расчет. Окисляемость (x), выраженную в мг/л O2 воды, вычисляют по формуле

где a - общий объем 0,01 н. раствора KMnO4, прибавленного в начале определения и израсходованного на титрование избытка щавелевой кислоты, мл;
b - объем 0,01 н. раствора KMnO4, израсходованного на титрование 10 мл щавелевой кислоты, мл;
k - поправочный коэффициент для приведения концентрации раствора KMnO4 к точно 0,01 н.;
V - объем анализируемой воды, взятой для определения, мл;
0,08 - эквивалент кислорода в мг на 1 мл 0,01 н. раствора KMnO4.
Окисляемость можно выражать также в миллиграммах KMnO4 на 1 л исследуемой воды.
В этом случае в расчет вместо 0,08 подставляют 0,32 - эквивалент KMnO4 в мг на 1 мл 0,01 н. раствора KMnO4.
В случае разбавления пробы дистиллированной водой формула для расчета окисляемости будет иметь следующий вид:
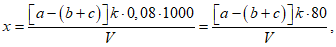
где c - объем 0,01 н. раствора KMnO4, требуемого для окисления того количества дистиллированной воды, которое было израсходовано для разбавления анализируемой воды, мл.
Определение методом Шульце-Паппа
11.7. Этот способ применяют для проб с содержанием хлоридов, превышающим 300 мг/л. Окисление проводят в щелочной среде; концентрации применяемых реактивов и ход определения, как при методе Кубеля.
Мешающие влияния
11.8. Мешающие влияния сероводорода, сульфидов, нитритов и двухвалентного железа устраняются вычитанием из полученного значения окисляемости отдельно определенных концентраций указанных веществ, пересчитанных на миллиграммы O2.
Аппаратура
11.9. Аппаратура такая же, как и для определения методом Кубеля.
Реактивы
11.10. Едкий натр ч.д.а. - 33%-ный раствор.
Остальные реактивы те же, что и для определения методом Кубеля.
Ход определения
11.11. В колбу для кипячения помещают несколько стеклянных шариков и отмеряют 100 мл пробы (после двухчасового ее отстаивания) или меньшее количество, доведенное до 100 мл дистиллированной водой, прибавляют 10 мл 0,01 н. раствора перманганата калия и 0,5 мл раствора едкого натра. Консервированную пробу нейтрализуют.
Смесь тщательно перемешивают, быстро нагревают до кипения и равномерно кипятят 10 мин. После окончания кипения добавляют к пробе 5 мл серной кислоты, 10 мл 0,01 н. титрованного раствора щавелевой кислоты и титруют 0,01 н. раствором перманганата до розовой окраски. Температура смеси при титровании не должна падать ниже 80 °C.
Расчет. Расчет производится так же, как и для метода Кубеля.
Определение четырехчасовым окислением
11.12. Четырехчасовое окисление применяют для определения окисляемости сточных вод, таких, как фекальные, или значительно загрязненных поверхностных вод. Определение отличается от остальных способов условиями проведения окисления. Перманганат, не израсходованный на окисление, определяют йодометрически. Нормальность применяемого раствора перманганата (0,125 н. или 0,0625 н.) выбирается в зависимости от степени загрязнения пробы воды.
Мешающие вещества
11.13. Мешающие вещества те же, что и при определении методом Кубеля. Необходимо, чтобы проба перед разбавлением была тщательно перемешана.
Аппаратура
11.14. Склянки емкостью около 100 мл.
Термостат (28 °C).
Реактивы
11.15. Перманганат калия, 0,125 н. раствор: 4 г KMnO4 растворяют в дистиллированной воде и доводят до 1 л. Раствор можно использовать не раньше чем через 2 - 3 недели.
Перманганат калия, 0,0625 н. раствор: 2 г KMnO4 растворяют в дистиллированной воде и доводят до 1 л. Раствор можно использовать не раньше чем через 2 - 3 недели.
Тиосульфат натрия, 0,05 н. раствор: 12,5 г Na2S2O3·5H2O ч.д.а. растворяют в дистиллированной воде, прибавляют 0,2 г Na2CO3 ч.д.а. и доводят до 1 л. Титр раствора определяют по стандартному раствору бихромата калия, как это описано на стр. 77.
Крахмал, индикатор, 0,5%-ный раствор: 5 г растворимого крахмала ч.д.а. смешивают с 50 мл дистиллированной воды и приливают к 950 мл кипящей дистиллированной воды.
Раствор консервируют прибавлением небольшого количества йодида ртути или другого консервирующего вещества, например амилового спирта, хлороформа или салициловой кислоты.
Йодид калия ч.д.а.
Серная кислота ч.д.а., разбавленная 1:3.
Ход определения
11.16. Сильно загрязненные пробы сточных вод перед определением разбавляют. Для проб сточных вод с неизвестной величиной окисляемости определение производят при двух или трех различных разбавлениях.
В две склянки, снабженные притертыми пробками, отмеривают пипеткой по 25 мл тщательно перемешанной пробы, 10 мл серной кислоты и в зависимости от загрязнения пробы прибавляют 10 мл 0,125 н. или 0,0625 н. раствора перманганата. Склянки закрывают пробкой и помещают в термостат при температуре 28 °C. После 4 ч в каждую склянку прибавляют около 0,2 г йодида калия, склянки закрывают снова пробкой, содержимое перемешивают и оставляют в темноте на 5 мин. Затем склянки открывают, пробку и горлышко ополаскивают дистиллированной водой в ту же склянку. Свободный йод титруют прямо в склянке 0,05 н. раствором тиосульфата натрия. В конце титрования прибавляют раствор крахмала.
Одновременно производят холостое определение с 25 мл дистиллированной воды, применяя тот же раствор перманганата и в таких же условиях, как и при анализе пробы сточной воды.
Правильным определением считается такое, при котором расход перманганата на титрование разбавленных проб составляет от 10 до 50% прибавленного количества перманганата.
Расчет. Окисляемость в мг/л вычисляют по формуле

где a - расход 0,05 н. раствора тиосульфата натрия на холостое определение, мл;
b - средний расход 0,05 н. раствора тиосульфата натрия на определение в двух параллельных пробах, мл;
k - поправка к нормальности раствора тиосульфата натрия;
V - количество пробы, взятое для определения, мл;
8 - эквивалент кислорода.
Бихроматный метод
11.17. Бихромат калия окисляет при кипячении в сернокислой среде большинство органических веществ, присутствующих в воде: избыток бихромата определяют титрованием. Для повышения полноты окисления некоторых веществ прибавляется сульфат серебра в качестве катализатора.
Использование двух концентраций (0,25 н. и 0,05 н.) бихромата калия позволяет определять окисляемость с точностью до 2 или 10 мг при концентрации от десятков до сотен мг/л O2. Верхний предел определяемой окисляемости после соответствующего разбавления пробы - 100 000 мг/л O2. Величины окисляемости, определяемые при применении 0,05 н. раствора бихромата, в некоторых случаях оказываются ниже, чем при титровании 0,26 н. раствором, не более чем на 10%.
Мешающие влияния
11.18. Мешающие влияния хлоридов (а также йодидов и бромидов в случае их присутствия) частично устраняются применением катализатора.
При работе с 0,05 н. раствором бихромата даже незначительные загрязнения посуды и холодильника следами органических веществ вызывают грубые ошибки определения. Ниже приводятся четыре варианта определения при наличии в воде: а) легко окисляемых веществ; б) трудно окисляемых веществ; в) сульфидов; г) нелетучих органических веществ.
Вариант А. Определение в присутствии
легко окисляемых веществ
Аппаратура
11.19. Колбы со шлифами, круглодонные, емкостью 250 мл.
Обратный холодильник.
Реактивы
11.20. Бихромат калия, 0,25 н. основной раствор; 12,258 г K2Cr2O7 ч.д.а., высушенного 2 ч при 105 °C, растворяют в дистиллированной воде и доводят объем при 20 °C до 1 л.
Бихромат калия 0,05 н. раствор: 50 мл 0,25 н. бихромата разбавляют дистиллированной водой при 20 °C до 1 л.
Серная кислота ч.д.а., концентрированная (96%-ная).
Сульфат серебра ч.д.а.
Соль Мора [сульфат железа (II) и аммония], 0,25 н. раствор; 98 г Fe(NH2)2(SO4)2·6H2O ч.д.а. растворяют в дистиллированной воде. Прибавляют 20 мл концентрированной серной кислоты и после охлаждения доводят объем до 1 л дистиллированной водой. Титр раствора устанавливают для каждой серии определений; 0,25 мл 0,25 н. раствора бихромата разбавляют дистиллированной водой приблизительно до 250 мл. Прибавляют 20 мл концентрированной серной кислоты и после охлаждения титруют раствором соли Мора при добавлении 3 - 4 капель ферроина или дифениламина (10 - 15 капель раствора фенилантраниловой кислоты).
Соль Мора, 0,05 н. раствор; приготовляют разбавлением 0,25 н. раствора. Титр устанавливается так же, как и 0,25 н. раствора.
Индикатор, применяют один из следующих растворов:
а) ферроин; 1,485 г моногидрата 1, 10-фенантралина и 0,695 г FeSO4·7H2O ч.д.а. растворяют в дистиллированной воде и доводят объем до 100 мл;
б) дифениламин, раствор; 0,5 г дифениламина растворяют в 100 мл серной кислоты (пл. 1,84) и вливают в 20 мл дистиллированной воды;
в) N-фенилантраниловая кислота; 0,25 г кислоты растворяют в 12 мл 0,1 н. раствора едкого натра и разбавляют дистиллированной водой до 250 мл.
Ход определения
11.21. При предполагаемой окисляемости исследуемой пробы ниже 50 мг/л используют для окисления 0,05 н. раствор бихромата, в остальных случаях применяют 0,25 н. раствор. Сильно загрязненные воды разбавляют перед определением так, чтобы расход бихромата составлял не более 50% прибавленного его количества. Пробы перед разбавлением перемешивают.
Пробу в 20 мл или меньший ее объем, доведенный дистиллированной водой до 20 мл, помещают в колбу со шлифом для кипячения. Прибавляют 10 мл 0,25 н. или 0,05 н. раствора бихромата, 0,4 г сульфата серебра и стеклянные шарики или кусок пемзы. Смесь перемешивают и к ней осторожно приливают 30 мл концентрированной серной кислоты, затем присоединяют обратный холодильник и равномерно кипятят 2 ч. После охлаждения отсоединяют холодильник, прибавляют в колбу 100 мл дистиллированной воды, смесь вновь охлаждают, прибавляют 3 - 4 капли раствора ферроина или дифениламина (10 - 15 капель раствора N-фенилантраниловой кислоты). Избыток непрореагировавшего бихромата титруют раствором соли Мора соответствующей концентрации до перехода окраски.
Таким же образом проводят холостой опыт с 20 мл дистиллированной воды.
Расчет. Окисляемость бихроматным методом, которую часто называют химическим поглощением кислорода (ХПК), выраженную числом миллиграммов O2/л воды, вычисляют по формуле

где a - расход титрованного раствора соли Мора на "холостой" опыт, мл;
b - расход титрованного раствора соли Мора на титрование пробы, мл;
k - поправка к нормальности раствора соли Мора;
F - пересчетный коэффициент; F = 400 для 0,05 н. раствора и F = 2000 для 0,25 н. раствора;
V - объем анализируемой воды, мл.
Если анализируемая вода содержит менее 100 мг/л хлоридов и в ней присутствуют лишь легко окисляемые органические вещества, то можно провести определение без добавления катализатора - сульфата серебра. Хлорид-ионы окисляются количественно до свободного хлора; из полученного результата надо вычесть поправку: на 1 мг хлорид-ионов расходуется 0,23 мг кислорода.
Вариант Б. Определение в присутствии
трудно окисляемых веществ
11.22. Если проба содержит органические вещества, окисляющиеся достаточно полно только в присутствии катализатора, рекомендуется в пробу вводить сульфат серебра. Если проба помимо органических веществ содержит хлориды, в концентрации более 100 мг/л следует прибавить сульфат ртути из расчета 15 мг сульфата ртути на каждый миллиграмм хлорид-иона.
Образуется растворимый, но очень мало диссоциированный хлорид ртути, который при избытке ионов ртути достаточно устойчив даже в присутствии концентрированной серной кислоты и бихромата.
Ход определения
11.23. К 20 мл пробы (или меньшему ее объему, разбавленному дистиллированной водой до 20 мл), содержащей во взятом объеме не более 40 мг хлорид-ионов, прибавляют 1 г сульфата ртути, 5 мл концентрированной серной кислоты для растворения соли ртути, 10 мл титрованного раствора бихромата калия, осторожно приливают 30 мл концентрированной серной кислоты, всыпают 0,4 г сульфата серебра и кипятят при слабом кипении в течение 2 ч. Заканчивают определение и ведут расчет, как описано выше.
Вариант В. Определение в присутствии сульфидов
11.24. Если в исследуемой воде присутствуют сульфиды, то при добавлении соли ртути выпадает черный осадок сульфида ртути, не растворяющийся при дальнейшей обработке.
В этих случаях рекомендуется изменить порядок введения реактивов по сравнению с описанным выше. К 20 мл исследуемой воды прибавляют 10 мл раствора бихромата, затем вливают 5 мл концентрированной серной кислоты и дают постоять 10 - 20 мин при комнатной температуре для окисления легкоокисляющихся веществ, в том числе и сернистых соединений. Затем прибавляют 1 г сульфата ртути, вводят 30 мл концентрированной серной кислоты, 0,4 г сульфата серебра и продолжают, как описано выше (стр. 65).
Вариант Г. Определение в присутствии
нелетучих органических веществ
11.25. Для определения окисляемости природных вод, содержащих нелетучие органические вещества (гуминовые соединения, обусловливающие цветность воды и др.), можно пользоваться методом бихроматной окисляемости с предварительным выпариванием воды досуха.
Аппаратура
11.26. Колбы плоскодонные емкостью 100 - 150 мл, предназначенные для выпаривания исследуемой воды.
Колпачки-холодильники.
Реактивы
11.27. Сернохромовая смесь, 0,2 н. раствор: 9,8 г K2Cr2O7 ч.д.а., высушенного в течение 2 ч при 105 °C, растворяют в 500 мл дистиллированной воды и затем прибавляют 500 мл H2SO4 ч.д.а. (пл. 1,84).
Соль Мора, 0,1 н. раствор: 39,2 г Fe(NH4)2·(SO4)2·H2O ч.д.а. растворяют в 1 л дистиллированной воды, содержащей 20 мл концентрированной H2SO4. Нормальность раствора соли Мора устанавливается по 0,1 н. раствору перманганата калия.
Индикаторы - ферроин, дифениламин или N-фенилантраниловая кислота. Приготовление см. стр. 65.
Ортофосфорная кислота, ч.д.а.
Ход определения
11.28. 50 - 250 мл исследуемой воды (в зависимости от содержания органических веществ) выпаривают в конических колбах на водяной бане досуха.
К сухому осадку в колбах приливают по 10 мл 0,2 н. сернохромовой смеси. Для отмеривания смеси пользуются пипеткой емкостью 10 мл. Время стекания со стенок должно составлять 1 мин (учитывают секундомером).
В качестве катализатора прибавляют 100 мг сульфата серебра. Содержимое колб осторожно перемешивают. Затем колбы помещают на нагретую до 180 - 200 °C (но не больше) песчаную баню или на горячую электроплитку с асбестовой сеткой, где их нагревают до кипения и кипятят в течение 5 мин. Кипение раствора должно быть равномерным.
Для предотвращения упаривания жидкости горло колб закрывают колпачками-холодильниками. Если во время кипения раствор принимает зеленую окраску, то для анализа следует взять меньше исследуемой воды.
Производят "холостое" определение. Кипятят 10 мл сернохромовой смеси с сульфатом серебра одновременно с пробами исследуемой воды и оттитровывают солью Мора.
После кипячения колбы охлаждают в течение 20 - 30 мин, затем нижнюю часть колпачков-холодильников и стенки колб обмывают небольшими порциями дистиллированной воды и содержимое переводят без потерь в коническую колбу емкостью 250 - 300 мл и доводят объем жидкости дистиллированной водой до 100 - 150 мл.
Для устранения влияния ионов окисного железа и получения перехода окраски индикатора более резким в колбы добавляют 2,5 мл фосфорной кислоты, и избыток сернохромовой смеси титруют 0,1 н. раствором соли Мора в присутствии одного из указанных индикаторов.
В конце титрования раствор соли Мора следует прибавлять по каплям.
Расчет. Окисляемость (ХПК) в мг/л O2 вычисляют по формуле
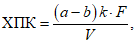
где a - расход титрованного раствора соли Мора на "холостое" титрование, мл;
b - расход титрованного раствора соли Мора на титрование, мл;
F - пересчетный коэффициент; F = 800 для 0,1 н. раствора соли Мора;
k - титр или поправка к нормальности раствора соли Мора;
V - объем анализируемой воды, мл.
Раскисляющее действие хлоридов (при наличии их в воде) устраняют внесением в упариваемую пробу сульфата серебра (в небольшом избытке от теоретически вычисленного по наличию хлоридов в исследуемом объеме). При наличии не более 50 мг хлоридов удается получить результаты с точностью +/- 3 + 4%. При большем содержании хлоридов ошибка возрастает.
Определение бихроматной окисляемости
при малом содержании органических веществ
11.29. Рассматриваемым методом можно определять бихроматную окисляемость только в природных мало загрязненных водах. Метод заключается в том, что пробу воды предварительно выпаривают в фарфоровой чашке досуха, сухой остаток растворяют в крепкой серной кислоте, содержащей бихромат калия в концентрации, соответствующей 0,5 н. раствору. Перед выпариванием в пробу добавляют 25 - 50 мг кальцинированной соды, чтобы не летели вещества, имеющие кислотные свойства.
Аппаратура
11.30. Бани водяные, электрические.
Плитки электрические.
Бани песчаные.
Бюретки со стеклянными кранами, емкостью 25 мл.
Капельницы.
Чашки фарфоровые, емкостью 250 - 300 мл.
Колбы конические, емкостью 400 - 500 мл.
Колбы круглодонные с пришлифованными обратными холодильниками.
Реактивы
11.31. Бихромата калия, 0,05 н. раствор в серной кислоте (пл. 1,84). Растворяют 2,4518 г K2Cr2O7, высушенного при 130 °C, в возможно малом количестве дистиллированной воды, осторожно добавляют серную кислоту до объема около 500 мл. После охлаждения смесь переносят в мерную колбу емкостью 1 л и доводят до метки концентрированной серной кислотой.
Соль Мора, 0,05 н. раствор: 19,2 г Fe(NH4)2·(SO4)2·H2O растворяют в 1 л дистиллированной воды, содержащей 20 мл концентрированной серной кислоты.
Серная кислота, разбавленная (1:4).
Сульфат серебра.
Сода кальцинированная.
Индикаторы: ферроин или N-фенилантраниловая кислота. Приготовление см. стр. 65.
Ход определения
11.32. Пробу воды в количестве 100 - 500 мл выпаривают досуха на водяной бане с добавлением 25 - 50 мг кальцинированной соды. К сухому остатку в чашке осторожно пипеткой приливают 25 мл 0,05 н. раствора бихромата калия. Для точного расхода количества окислителя во всех пробах придерживаются одинакового времени выливания содержимого из пипетки (1 мин с момента прекращения непрерывного вытекания). Сухое вещество в чашке оставляют с раствором бихромата калия на 3 - 5 мин и затем осторожно переносят в круглодонную колбу с обратным холодильником. Чашку три раза промывают серной кислотой (1:4), расходуя для этого 50 мл кислоты, и добавляют 100 мг сульфата серебра (катализатор) и кусочек пемзы для равномерного кипения. Колбу нагревают на плитке с асбестом или на песчаной бане в течение 30 мин, с момента начала кипения. После охлаждения пробу переносят в коническую колбу емкостью 750 мл, ополаскивают круглодонную колбу дистиллированной водой и доливают дистиллированной водой до 300 мл. Затем добавляют 3 - 5 капель раствора ферроина или 10 - 15 капель раствора N-фенилантраниловой кислоты. Избыток бихромата калия оттитровывают 0,05 н. раствором соли Мора до перехода окраски индикатора. Круглодонную колбу с холодильником (прибор Штеребингера) для определения ХПК предварительно обрабатывают хромовой смесью при кипячении примерно в течение 30 мин. Всю посуду перед употреблением следует хорошо промыть хромовой смесью и пропарить.
Расчет. Окисляемость бихроматным методом (ХПК) в мг/л O2 вычисляют по формуле
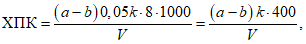
где a - объем раствора соли Мора, израсходованной на титрование в "холостом опыте", мл;
b - объем того же раствора, израсходованного на титрование пробы, мл;
0,05 - нормальность раствора соли Мора;
k - поправка к нормальности раствора соли Мора;
V - объем анализируемой воды, мл;
8 - грамм-эквивалент кислорода.
Общие положения
12.1. Вода, соприкасающаяся с воздухом, содержит кислород в равновесной концентрации, зависящей от атмосферного давления, температуры и содержания растворенных в воде солей. Значения равновесных концентраций кислорода в дистиллированной воде, которая при нормальном давлении соприкасается с воздухом, содержащим 20,9% кислорода, не содержащим CO2 и насыщенным водяным паром, приведены в табл. 8 (стр. 71).
Таблица 8
для вычисления насыщения кислородом
Температура, °C | Растворенный кислород, мг/л | |||||||||
0,0 | 0,1 | 0,2 | 0,3 | 0,4 | 0,5 | 0,6 | 0,7 | 0,8 | 0,9 | |
0 | 14,65 | 14,61 | 14,57 | 14,53 | 14,49 | 14,45 | 14,41 | 14,37 | 14,33 | 14,29 |
1 | 14,25 | 14,21 | 14,17 | 14,13 | 14,09 | 14,05 | 14,02 | 13,98 | 13,94 | 13,90 |
2 | 13,86 | 13,82 | 13,79 | 13,75 | 13,71 | 13,68 | 13,64 | 13,60 | 13,56 | 13,53 |
3 | 13,49 | 13,46 | 13,42 | 13,38 | 13,35 | 13,31 | 13,28 | 13,24 | 13,20 | 13,17 |
4 | 13,13 | 13,10 | 13,06 | 13,03 | 13,00 | 12,96 | 12,93 | 12,89 | 12,86 | 12,82 |
5 | 12,79 | 12,76 | 12,72 | 12,69 | 12,55 | 12,52 | 12,59 | 12,56 | 12,53 | 12,49 |
6 | 12,46 | 12,43 | 12,40 | 12,36 | 12,33 | 12,30 | 12,27 | 12,24 | 12,21 | 12,18 |
7 | 12,14 | 12,11 | 12,08 | 12,05 | 12,02 | 11,99 | 11,96 | 11,93 | 11,90 | 11,87 |
8 | 11,84 | 11,81 | 11,78 | 11,75 | 11,72 | 11,70 | 11,67 | 11,64 | 11,61 | 11,58 |
9 | 11,55 | 11,52 | 11,49 | 11,47 | 11,44 | 11,41 | 11,38 | 11,35 | 11,33 | 11,30 |
10 | 11,27 | 11,24 | 11,22 | 11,19 | 11,16 | 11,14 | 11,11 | 11,08 | 11,06 | 11,03 |
11 | 11,00 | 10,98 | 10,95 | 10,93 | 10,90 | 10,87 | 10,85 | 10,82 | 10,80 | 10,77 |
12 | 10,75 | 10,72 | 10,70 | 10,67 | 10,65 | 10,62 | 10,60 | 10,57 | 10,55 | 10,52 |
13 | 10,50 | 10,48 | 10,45 | 10,43 | 10,40 | 10,38 | 10,36 | 10,33 | 10,31 | 10,28 |
14 | 10,26 | 10,24 | 10,22 | 10,19 | 10,17 | 10,15 | 10,12 | 10,10 | 10,08 | 10,06 |
15 | 10,03 | 10,01 | 10,99 | 9,97 | 9,95 | 9,92 | 9,90 | 9,88 | 9,86 | 9,84 |
16 | 9,82 | 9,79 | 9,77 | 9,75 | 9,73 | 9,71 | 9,69 | 9,67 | 9,65 | 9,63 |
17 | 9,61 | 9,58 | 9,56 | 9,54 | 9,52 | 9,50 | 9,48 | 9,46 | 9,44 | 9,42 |
18 | 9,40 | 9,38 | 9,36 | 9,34 | 9,32 | 9,30 | 9,29 | 9,27 | 9,25 | 9,23 |
19 | 9,21 | 9,19 | 9,17 | 9,15 | 9,13 | 9,12 | 9,10 | 9,08 | 9,06 | 9,04 |
20 | 9,02 | 9,00 | 8,98 | 8,97 | 8,95 | 8,93 | 8,91 | 8,90 | 8,88 | 8,86 |
21 | 8,84 | 8,82 | 8,81 | 8,79 | 8,77 | 8,75 | 8,74 | 8,72 | 8,70 | 8,68 |
22 | 8,67 | 8,65 | 8,63 | 8,62 | 8,60 | 8,58 | 8,56 | 8,55 | 8,53 | 8,52 |
23 | 8,50 | 8,48 | 8,46 | 8,45 | 8,43 | 8,42 | 8,40 | 8,38 | 8,37 | 8,35 |
24 | 8,33 | 8,32 | 8,30 | 8,29 | 8,27 | 8,25 | 8,24 | 8,22 | 8,21 | 8,19 |
25 | 8,18 | 8,16 | 8,14 | 8,13 | 8,11 | 8,10 | 8,08 | 8,07 | 8,05 | 8,04 |
26 | 8,02 | 8,01 | 7,79 | 7,98 | 7,96 | 7,95 | 7,93 | 7,92 | 7,90 | 7,89 |
27 | 7,87 | 7,86 | 7,84 | 7,83 | 7,81 | 7,80 | 7,78 | 7,77 | 7,75 | 7,74 |
28 | 7,72 | 7,71 | 7,69 | 7,68 | 7,66 | 7,65 | 7,64 | 7,62 | 7,61 | 7,59 |
29 | 7,58 | 7,56 | 7,55 | 7,54 | 7,52 | 7,51 | 7,49 | 7,48 | 7,47 | 7,45 |
30 | 7,44 | 7,42 | 7,41 | 7,40 | 7,38 | 7,37 | 7,35 | 7,34 | 7,32 | 7,31 |
Отклонения действительной концентрации кислорода от равновесной вызываются: а) физическими влияниями, например резким изменением барометрического давления, изменением температуры воды, аэрацией воды в плотинах; б) физико-химическими и химическими влияниями, например, поглощением кислорода при электрокоррозии металлов и потреблением его на химическое окисление веществ, содержащихся в воде или соприкасающихся с ней; в) биохимическими влияниями, которые в естественных условиях преобладают, как, например, потреблением кислорода при аэробном микробиальном разложении органических веществ или, наоборот, выделением кислорода при поглощении CO2 организмами.
Для определения растворенного кислорода приводится йодометрический метод.
Определение кислорода основано на реакции растворенного кислорода с гидроокисью марганца (II) и на йодометрическом определении образовавшихся высших по степени окисления соединений марганца. Два изложенных ниже варианта определения отличаются лишь техникой выполнения. Вариант А пригоден для массовых определений, так как он проще; его можно применять только в тех случаях, когда осадок гидроокиси марганца был полностью отделен от раствора отстаиванием. Вариант Б применяется в том случае, когда отсутствует полное отделение осадка, а также при устранении некоторых мешающих влияний, как это указано ниже.
Точность определения растворенного кислорода обоими методами достигает для чистых вод +0,05 мг/л. При анализе загрязненных или сточных вод ошибка определения иногда превышает +/- 0,1 мг/л. Описанными методами можно определить кислород при концентрации его 0,2 - 0,3 мг/л. Для определения более низких концентраций необходимо применять специальный метод.
Пробы для определения кислорода отбирают в так называемые "кислородные" склянки (в исключительных случаях в бутыли емкостью 1 л при устранении мешающих влияний перед фиксацией). Во время отбора пробы в горло бутыли вставляют специальную насадку и бутыль погружают ниже уровня воды, где ее оставляют до полного наполнения. Насадку надо вынимать лишь под водой. При отборах проб воду следует наливать резиновой трубкой, опущенной на дно кислородной склянки, и после наполнения оставлять некоторое время в токе воды. Немедленно после отбора пробы кислород фиксируют (см. "Ход определения").
Результат определения кислорода выражают в миллиграммах на 1 л воды.
Мешающие влияния
12.2. При определении кислорода мешающие влияния оказывают любые изменения концентрации кислорода в пробе в период между отбором и фиксацией, а также изменение состава осадка гидроокиси марганца в период с момента ее образования до определения титрованием. Некоторые вещества, содержащиеся в пробе анализируемой воды, могут оказать мешающее действие тем, что понижают концентрацию выделившегося йода адсорбцией на своей поверхности или химической реакцией с йодом, или наоборот. Для устранения мешающих влияний приведены операции, которые частично меняют ход двух основных определений кислорода (варианты А и Б). Кроме того, приведены методы подготовки пробы, которые необходимо проводить перед определением кислорода по одному из вариантов.
Если не известен характер мешающих влияний, кислород определяется по схеме, описанной в подпунктах 2 и 5.
1) Изменение концентрации кислорода между отбором пробы и выпадением осадка гидроокиси марганца (II) чаще всего вызвано повышением температуры отобранной пробы, химическим или биохимическим потреблением или же образованием кислорода. Поэтому в пробе, отобранной для определения кислорода, его необходимо немедленно после отбора фиксировать прибавлением растворов сульфата марганца и едкого кали для перевода в осадок.
2) В полевых условиях при более продолжительном времени с момента фиксации кислорода до его титрования (более 1 суток) возникает опасность, что осадок гидроокиси марганца (II) начнет окисляться кислородом, который может проникнуть в кислородную склянку через шлиф. В таких случаях необходимо перевести гидроокиси марганца в карбонаты, которые более устойчивы и не окисляются растворенным кислородом. Кислород фиксируется обычно прибавлением раствора сульфата марганца (II) и едкого кали (без добавления йодида). После перемешивания и выпадения осадка гидроокиси марганца (II) кислородную склянку открывают, прибавляют примерно 3 г бикарбоната калия, вновь закрывают так, чтобы не было воздушных пузырьков, и содержимое хорошо перемешивают опрокидыванием склянки. По ходу определения содержимое кислородной склянки вместе с осадком переводят в колбу для титрования, а в кислородную склянку прибавляют 10 мл кислоты (для растворения), которой ополаскивают склянку. Кислоту переливают в колбу для титрования, кислородную склянку ополаскивают дистиллированной водой, которую также затем приливают в ту же колбу. После этого содержимое колбы перемешивают и по окончании выделения углекислого газа прибавляют 2 мл 15%-ного раствора йодида калия. После перемешивания выделившийся йод титруют раствором тиосульфата натрия, как описано ниже.
При анализе проб, у которых мешающие влияния должны быть устранены перед выделением гидроокисей марганца, необходимо выбрать ход определения так, чтобы концентрация кислорода в ней при обработке не изменялась.
3) Если в пробе имеется большое количество взвешенных веществ, необходимо в некоторых случаях их устранить перед фиксацией кислорода. Взвешенные вещества могут в кислой среде адсорбировать элементарный йод и тем самым снизить результат определения; кроме того, они могут мешать визуальному определению точки перехода окраски при титровании. Если нет опасения в снижении концентрации кислорода в результате микробиальных процессов, пробу осветляют гидроокисью алюминия. Стеклянную бутылку емкостью 1 л с притертой пробкой наполняют доверху пробой с помощью специальной насадки. Пипеткой прибавляют 10 мл 10%-ного раствора сульфата алюминия и калия (10 г KAl(SO4)2·12H2O растворяют в дистиллированной воде и доводят до 100 мл) и 2 мл концентрированного раствора аммиака. Бутылку закрывают притертой пробкой так, чтобы в ней не оставалось пузырьков воздуха, и опрокидыванием бутылки перемешивают содержимое примерно 1 мин. Образовавшемуся осадку дают отстояться. Пробу не следует оставлять непосредственно на прямом солнечном свету или вблизи какого-либо источника тепла. Примерно через 10 мин прозрачную пробу над осадком переводят сифоном в кислородную склянку. Во избежание обогащения пробы кислородом воздуха конец сифона помещают на дно кислородной склянки и оставляют последнюю медленно наполняться раствором до тех пор, пока не перетечет по крайней мере одна треть содержимого бутылки. Затем сифон вынимают и кислород фиксируют, как описано в вариантах А и Б.
4) Пробы, содержащие плохо осаждающиеся взвешенные вещества, которые могут вызвать снижение концентрации кислорода вследствие интенсивной жизнедеятельности микроорганизмов, необходимо осветлять при одновременном прибавлении токсичного вещества. Пробу отбирают, как и в предыдущем случае, в бутылку емкостью 1 л, снабженную притертой пробкой. Сразу после отбора к ней прибавляют 10 мл раствора сульфаминовой кислоты и хлорида ртути (II) (32 г сульфаминовой кислоты NH2SO3H растворяют в 450 мл дистиллированной воды; 54 г хлорида ртути (II) растворяют при нагревании в 450 мл дистиллированной воды; оба раствора смешивают и доводят дистиллированной водой до 1 л) и 10 мл 10%-ного раствора сульфата алюминия и калия. Реактивы можно внести в бутылку и до отбора пробы. После наполнения бутылку закрывают притертой пробкой, не оставляя пузырьков воздуха, и содержимое хорошо перемешивают опрокидыванием бутылки. Затем прибавляют 5 мл 2 н. раствора едкого натра (80 г NaOH ч.д.а. растворяют в дистиллированной воде и доводят до 1 л), бутылку закрывают и содержимое опрокидыванием бутылки перемешивают примерно 1 мин. Далее определение продолжают согласно п. 3 (см. стр. 72). Кислород определяется по варианту Б.
5) Пробы, содержащие хорошо осаждающиеся взвешенные вещества, которые могут снизить концентрацию растворенного кислорода за счет интенсивной жизнедеятельности микроорганизмов, подготавливают к определению прибавлением раствора сульфата меди (II), сульфаминовой кислоты и отстаиванием. Такую подготовку применяют, например, при анализе воды, содержащей активный ил. Пробу отбирают специальной насадкой в бутылку емкостью 1 л с притертой пробкой. До отбора или сразу после отбора в бутылку прибавляют 10 мл раствора сульфата меди (II) и сульфаминовой кислоты (32 г сульфаминовой кислоты NH2SO3H растворяют в 450 мл дистиллированной воды; 50 г CuSO4·5H2O растворяют в 450 мл дистиллированной воды; оба раствора смешивают, прибавляют 26 мл уксусной кислоты и доводят до 1 л). Бутылку закрывают пробкой так, чтобы под ней не было пузырьков воздуха, и содержимое перемешивают несколько раз, переворачивая бутылку. Время отстаивания зависит в данном случае от свойств взвешенных веществ, однако оно должно быть по возможности небольшим. Затем пробу обрабатывают способом, описанным в п. 3 настоящего раздела.
Определению кислорода мешают вещества, которые выделяют йод или в кислой среде реагируют с йодом. Окисление йодида до йода, приводящее к положительной ошибке определения, вызывают, например, свободный хлор, хлорамин, двуокись хлора, бихромат, перманганат, железо (III) и перекиси. Восстановление йода в йодид, приводящее к отрицательной ошибке определения, вызывают, например, сульфиты и сульфиды. Некоторые органические соединения приводят к отрицательной ошибке при определении кислорода тем, что они окисляются выделенным йодом или реагируют с ним (реакции присоединения и замещения). Некоторые минеральные вещества, например, железо (II), и некоторые органические соединения влияют на определение, так как в щелочной среде легко окисляются растворенным кислородом. Указанные влияния устраняют следующим образом.
6) Для проб, содержащих органические вещества, легко окисляемые в щелочной среде кислородом и окисляемые йодом в кислой среде, наилучшим является ход определения по Терио (Theriault). Отличие этого определения от варианта Б состоит в том, что после фиксации кислорода осадок оставляют выпадать только до тех пор (достаточно 2 - 3 мин отстаивания), пока под горлышком кислородной склянки не образуется прозрачный слой; тотчас же прибавляют кислоту, содержимое кислородной склянки быстро переводят в колбу для титрования и немедленно титруют.
7) Если требуется повышенная точность определения, мешающее влияние органических и минеральных веществ учитывают проводя холостой опыт. Пробу, предназначенную для холостого опыта, отбирают одновременно с анализируемой пробой. В пробе воды фиксируют кислород прибавлением 2 мл раствора сульфата марганца (II) и 2 мл раствора едкого кали с добавлением йодида и азида (приготовление см. стр. 76). В кислородную склянку при проведении холостого опыта прибавляют только 2 мл раствора едкого кали с добавлением йодида и азида. Затем анализируемую пробу и раствор холостого опыта обрабатывают в соответствии с вариантом Б, причем соблюдают указанные условия и в отношении времени. В случае выделения в холостой пробе йода после подкисления и прибавления раствора йодида его титруют тиосульфатом. Расход тиосульфата пересчитывают на кислород и выражают в миллиграммах на 1 л, так же как и при определении кислорода (из объема кислородной склянки вычитают 2 мл). Результат холостого опыта вычитают из результата определения.
Если после подкисления в холостой пробе йод не выделяется, в колбу для титрования прибавляют 25 мл 0,01 н. раствора йода (приготовление см. стр. 208) и оставляют в покое столько же времени, сколько и анализируемую пробу. Избыток йода титруют раствором тиосульфата. Одновременно определяют количество прибавленного йода титрованием 25 мл раствора йода, прибавленного к 100 мл дистиллированной воды, подкисленной 5 мл кислоты (для растворения). Разность между результатами обоих титрований показывает количество йода, восстановленного веществами, мешающими определению. Эту разность пересчитывают на мг/л O2 и учитывают при расчете результатов определения.
8) Если проба содержит минеральные или органические вещества, которые в сильнощелочной среде находятся в растворенной форме и мешают только при йодометрическом титровании, их влияние можно устранить отделением осадка гидроокисей марганца. Осадок после стабилизации в соответствии с п. 2 отделяют фильтрованием: содержимое кислородной склянки фильтруют через воронку с фильтрующей стеклянной пластинкой. Кислородную склянку и оставшийся в ней осадок ополаскивают дистиллированной водой. Фильтрат отбрасывают. Затем в кислородную склянку прибавляют 2 мл 15%-ного раствора йодида калия и 5 мл кислоты (для растворения). Обмывают этой смесью стенки кислородной склянки и постепенно переносят ее на стеклянный фильтр, чтобы осадок на фильтре растворился. Раствор собирают в колбу для титрования. Кислородную склянку и фильтр промывают дистиллированной водой. Объем соединенных фильтратов доводят примерно до 100 мл и титруют раствором тиосульфата.
9) Железо (III) в кислой среде выделяет йод из раствора йодида. Эта реакция обратима, протекает медленно, и количество выделенного йода зависит от концентрации веществ в растворе, условий протекания реакции, а также от времени. Присутствие железа (III) приводит к положительной ошибке определения и мешает обесцвечиванию раствора крахмала (синяя окраска возобновляется). При концентрации железа (III) ниже 1 мг/л мешающим влиянием его можно пренебречь, при более высоких концентрациях железа (III) (до 200 мг/л) его мешающее влияние устраняется прибавлением 1 мл 40%-ного раствора фторида калия (40 г KF·2H2O растворяют в дистиллированной воде и доводят до 100 мл) перед растворением осадка гидроокисей кислотой. Точное определение выделенного йода производят немедленно после подкисления пробы быстрым титрованием ее до первого обесцвечивания раствора крахмала.
10) Железо (II), присутствующее в пробе, при фиксации кислорода выделяется в виде гидроокиси железа (II), которая реагирует с растворенным кислородом быстрее, чем гидроокись марганца (II). Тем самым теряется часть растворенного кислорода. Подготовка пробы заключается в окислении железа (II) до железа (III) перманганатом калия: к пробе, отобранной в кислородную склянку, прибавляют точно 0,7 мл концентрированной серной кислоты, 1 мл раствора перманганата калия (6,3 г на 1 л) и 1 мл 40%-ного раствора фторида калия. Кислородную склянку закрывают и содержимое ее хорошо перемешивают перевертыванием. Фиолетовая окраска пробы должна сохраняться по крайней мере 5 мин. В случае более быстрого обесцвечивания пробы прибавляют еще 1 мл раствора перманганата калия. Избыток его удаляют прибавлением 2%-ного раствора оксалата калия (2 г K2C2O4·H2O на 100 мл) в количестве, точно необходимом для обесцвечивания пробы (0,5 - 1 мл). Избыток оксалата не должен превышать 0,5 мл, потому что повышенное его количество может вызвать отрицательную ошибку определения. Обесцвечивание пробы проводят в темноте в течение 2 - 10 мин. После полного обесцвечивания пробы в ней фиксируют кислород. Для фиксации кислорода прибавляют 2 мл раствора, хлорида марганца (II) и 3 мл раствора едкого кали. После подкисления пробу сразу же титруют до начала обесцвечивания раствора крахмала. При расчете из объема кислородной склянки вычитают объем всех прибавленных растворов, т.е. серной кислоты, перманганата калия, фторида калия, оксалата калия и растворов для осаждения.
11) Нитриты в кислой среде, действуя как катализатор, способствуют окислению йодидов до йода кислородом воздуха, что приводит к повышенному расходу тиосульфата и мешает окончанию титрования, так как окраска индикатора через некоторое время восстанавливается.
Это мешающее влияние устраняют в обоих вариантах (А и Б) тем, что в растворы едкого кали вводят азид натрия.
Аппаратура
12.3. Кислородные склянки емкостью 250 - 300 мл с косо срезанной притертой пробкой, калиброванные с точностью до 0,1 мл. Проверка емкости или калибровка производится взвешиванием. Объем кислородной склянки определяют вычитанием веса пустой склянки с пробкой из веса склянки, наполненной дистиллированной водой при 20 °C и закрытой пробкой. Перед взвешиванием необходимо тщательно высушить склянку, в особенности ее горло.
Бутылки для отбора проб емкостью 1 л, снабженные притертыми пробками.
Насадка для отбора проб.
Пипетки для прибавления фиксирующих растворов, цилиндрические с оттянутым носиком.
Сифон.
Реактивы
12.4. Сульфат марганца (II), раствор: 400 г MnSO4·2H2O (480 г MnSO4·4H2O или 364 г MnSO4·H2O) растворяют в дистиллированной воде и доводят объем до 1 л. Фильтруют через бумажный фильтр.
Едкое кали с азидом, раствор: 700 г KOH растворяют в 700 мл дистиллированной воды; 10 г азида натрия NaN3 растворяют в 40 мл дистиллированной воды; оба раствора смешивают и доводят до 1 л. Если раствор не прозрачен, ему дают отстояться и затем сифонируют.
Едкое кали с йодидом и азидом натрия, раствор: 700 г KOH и 150 г KJ растворяют в 700 мл дистиллированной воды; 10 г NaN3 растворяют в 40 мл дистиллированной воды; оба раствора смешивают и доводят до 1 л.
Если раствор не прозрачен, ему дают отстояться и сифонируют (2 мл раствора помещают в колбу, содержащую 100 мл дистиллированной воды и 10 мл разбавленной (1:4) серной кислоты; после прибавления 5 мл раствора крахмала не должна появиться окраска).
Серная кислота, ч.д.а., разбавленная (1:4).
Йодид калия, 15%-ный раствор: 15 г KJ ч.д.а. растворяют в дистиллированной воде, прибавляют 1 мл 1 н. раствора NaOH и доводят до 100 мл (2 мл раствора, помещенного в колбу, содержащую 100 мл дистиллированной воды и 5 мл разбавленной (1:4) серной кислоты; после прибавления 5 мл раствора крахмала не должна появиться окраска).
Тиосульфат натрия, 0,05 н. раствор: 12,4 г Na2S2O3·5H2O ч.д.а. и 0,2 г Na2CO3 ч.д.а., растворяют в дистиллированной воде и доводят до 1 л. Для установки титра в коническую колбу, снабженную притертой пробкой, приливают примерно 100 мл дистиллированной воды, прибавляют 10 мл 15%-ного раствора йодида калия, 5 мл разбавленной (1:4) серной кислоты и 20 мл 0,05 н. раствора бихромата калия. После перемешивания раствор оставляют стоять 5 мин в темноте и затем титруют раствором тиосульфата с 1 - 2 мл раствора крахмала в качестве индикатора. Титр проверяют не реже чем 1 раз в неделю. Можно применять титрованные растворы тиосульфата и другой нормальности, например 0,025 н. или 0,01 н. Их приготавливают аналогичным способом и устанавливают их титр, как описано выше. Эти растворы можно приготовить также соответствующим разбавлением более концентрированного запасного раствора, например 0,1 н.
Бихромат калия, 0,05 н. (0,02 н. и 0,01 н.) основной раствор: 2,4518 г K2Cr2O7 (0,9807 г при приготовлении 0,02 н. раствора и 0,4904 г при приготовлении 0,01 н. раствора), высушенного при 105 °C, растворяют в дистиллированной воде и доводят при 20 °C до 1 л.
Крахмал (индикаторный) 0,5%-ный, см. стр. 63.
Вместо раствора сульфата марганца можно применять также раствор хлорида марганца (525 г MnCl2·4H2O ч.д.а. растворяют в дистиллированной воде и доводят до 1 л). Для растворения осадка гидроокисей марганца в этом случае применяют разбавленную соляную кислоту (1:1).
Вместо едкого кали можно применить едкий натр (для приготовления раствора вместо 700 г KOH отвешивают 500 г NaOH; дальнейшее приготовление раствора остается без изменений).
Количества прибавляемых реактивов при этих заменах остаются без изменений, как указано в ходе определения.
Ход определения (Метод Винклера)
12.5. Вариант А. В кислородную склянку, заполненную доверху пробой, прибавляют 2 мл раствора сульфата марганца следующим способом: наполненную пипетку погружают до самого дна кислородной склянки, затем открывают верхний конец и пипетку медленно вынимают. Другой пипеткой прибавляют к пробе 2 мл раствора едкого кали с азидом. Кончик пипетки при этом опускают только под уровень пробы в горле кислородной склянки. Склянку осторожно закрывают так, чтобы под пробкой не образовались пузырьки воздуха. Горло ополаскивают водой и содержимое хорошо перемешивают переворачиванием склянки до образования хлопьевидного, хорошо выпадающего осадка.
Осадку дают собраться на дне склянки. Прозрачный раствор над осадком сифонируют. Конец сифона погружают до половины высоты кислородной склянки. Раствор можно слить сифоном или же присоединить сифон к водоструйному насосу и отсосать. Отсасывание должно продолжаться 15 - 20 сек (более быстрое отсасывание может увлечь осадок).
Сразу после отсасывания или сифонирования прозрачной жидкости над осадком в кислородную склянку по стенке приливают 5 мл разбавленной серной кислоты и перемешивают. Затем прибавляют 2 мл раствора йодида калия и содержимое вновь перемешивают. По истечении 5 мин выделившийся йод титруют непосредственно в кислородной склянке раствором тиосульфата до светло-желтой окраски.
Потом прибавляют раствор крахмала и продолжают титровать до обесцвечивания. Индикатор обычно прибавляют в количестве 1 - 2 мл в зависимости от качества применяемого крахмала.
12.6. Вариант. Б. Кислород фиксируют так же, как в варианте А. Приливают 2 мл раствора сульфата марганца и 2 мл раствора едкого кали с азидом или раствора едкого кали с йодидом калия и азидом.
После выпадения осадка гидроокисей марганца кислородную склянку открывают и пипеткой с широким концом, опущенным под уровень пробы, прибавляют 10 мл разбавленной серной кислоты. Кислородную склянку вновь закрывают и перевертыванием перемешивают ее содержимое. Затем содержимое кислородной склянки переливают в колбу для титрования и ополаскивают склянку дистиллированной водой, сливая ее в ту же колбу. Если для фиксации кислорода применялся раствор едкого кали без йодида, в колбу прибавляют еще 2 мл раствора йодида калия. По истечении 5 мин йод титруют раствором тиосульфата, как в варианте А.
Расчет. Содержание растворенного кислорода x в мг/л вычисляют по формуле

где a - расход титрованного раствора тиосульфата, мл;
k - поправка к нормальности раствора тиосульфата;
0,05 - нормальность раствора тиосульфата, примененного для титрования;
V1 - объем кислородной склянки, мл;
V2 - общий объем реактивов, прибавленных в кислородную склянку при фиксации кислорода, а в некоторых случаях и при предварительной подготовке пробы, мл;
8 - эквивалент кислорода.
Процент насыщения растворенным кислородом (y) вычисляют по формуле

где c1 - найденная концентрация кислорода, мг/л;
c2 - равновесная концентрация кислорода, взятая из табл. 8 для температуры воды, измеренной при отборе пробы, с возможной поправкой на атмосферное давление и на содержание растворенных солей, мг/л.
Округление результатов
Результаты округляют до 0,1 мг или до целых процентов.
Приведенные в табл. 8 равновесные концентрации даны для атмосферного давления 760 мм рт. ст. Для других атмосферных давлений равновесные концентрации вычисляют по формуле

где c1 - искомая величина равновесной концентрации кислорода при давлении P, мг/л;
c - величина равновесной концентрации кислорода, найденная по таблице для данной температуры воды, мг/л;
P - атмосферное давление, мм рт. ст.
Влияние солей, растворенных в воде, сказывается в уменьшении растворимости кислорода.
Растворимость уменьшается на каждые 1000 мг солей в 1 л воды на следующую величину:
Температура, °C | 0 | 10 | 20 | 30 |
Уменьшение растворимости, мг/л | 0,08405 | 0,06217 | 0,04777 | 0,04085 |
Определение кислорода по Винклеру
с предварительным окислением пробы воды для устранения
мешающих влияний восстановителей
Аппаратура
12.7. Кислородные склянки объемом 110 - 130 мл с пришлифованными пробками.
Бюретка, емкостью 25 мл.
Реактивы
12.8. Серная кислота 1:4.
Сульфат натрия. Раствор 250 г Na2SO4·10H2O растворяют при нагревании в 1 л дистиллированной воды.
Гипохлорит натрия, раствор 200 мл раствора сульфата натрия смешивают с 30 мл 3%-ного раствора NaClO. 1 мл разбавленного раствора NaClO должен соответствовать 8 - 10 мл раствора 0,01 н. раствора Na2S2O3.
Раствор NaClO хранится в темной склянке. Он должен быть защищен от прямых солнечных лучей. В разбавленном растворе содержание активного хлора убывает медленно. Этот раствор время от времени следует контролировать и, если нужно, укреплять концентрированным раствором NaClO.
Роданид калия, раствор 2 г KCNS растворяют в 200 мл раствора сульфата натрия.
Остальные реактивы см. стр. 76.
Ход анализа
12.9. В каждую кислородную склянку, наполненную пробой воды до краев, приливают из пипетки сначала 0,5 мл H2SO4 (1:4), затем 0,5 мл раствора NaClO. После этого склянку закрывают пробкой, взбалтывают так, чтобы в склянке не было пузырьков воздуха, и ставят на 30 мин в темное место. Затем в каждую кислородную склянку для устранения избытка NaClO добавляют по 1,0 мл раствора KCNS. Склянку закрывают пробкой и взбалтывают содержимое так, чтобы не попали пузырьки воздуха, и опять ставят на 10 мин. Затем в каждую склянку приливают по 2,0 мл раствора сульфата марганца и 1 мл раствора едкого натра, содержащего йодид и азид, закрывают склянку и перемешивают. Далее поступают как в обычном методе Винклера (стр. 77).
Расчет. 1 мл 0,01 н. раствора Na2S2O3 соответствует 0,08 мг O2. Содержание кислорода (x) в мг/л вычисляют по формуле

где a - количество 0,01 н. раствора Na2S2O3, израсходованного на титрование, мл;
k - поправка для приведения нормальности раствора Na2S2O3 к точно 0,01 н.;
V1 - объем кислородной склянки, мл;
V2 - общий объем реактивов, прибавленных в кислородную склянку при фиксации кислорода, мл;
8 - эквивалент кислорода.
При применении указанных количеств реактивов отнимают 3,5 мл от объема склянки. Сюда входят: 0,5 мл H2SO4 (1:4); 0,5 мл раствора NaClO, 1,0 мл раствора KCNS; 0,5 мл раствора MnCl2, 1,0 мл раствора едкого натра, содержащего йодид и азид.
Результаты округляются до 0,1 мг/л O2.
В ЧИСТЫХ И ЗАГРЯЗНЕННЫХ ПРИРОДНЫХ ВОДАХ
Общие положения
13.1. Методом биохимического потребления кислорода определяют количество растворенного кислорода (в миллиграммах на 1 л воды), которое требуется для окисления органического вещества аэробными бактериями.
Расход кислорода на нитрификацию, которая обычно является стадией, последующей за окислением органического вещества, не входит в величину БПК.
Определить истинное значение полного БПК сложно, так как при малом содержании в пробе органических веществ одновременно с их окислением идет процесс нитрификации.
Предлагаемый метод определения БПК до появления азота нитритов в количестве 0,1 мг/л с достаточной полнотой характеризует ход процесса биохимического окисления.
Время, нужное для полной минерализации органического вещества, зависит при прочих равных условиях от природы органических веществ, содержащихся в испытуемой жидкости.
Определение БПК производится в натуральной или в соответственно разбавленной пробе воды по разнице между содержанием растворенного кислорода в склянке в момент постановки опыта и после определенного периода инкубации. Пробы воды для определения БПК не консервируют, а анализируют сразу после отбора.
Определение БПК в натуральной пробе (без разбавления)
13.2. Для определения БПК воды с предполагаемым потреблением кислорода до 6 мг/л пробу воды отбирают в бутыль емкостью 2 л. Температура воды должна быть 20 +/- 1 °C; если температура иная, то в лаборатории воду подогревают на водяной бане или охлаждают до требуемой температуры и сильно встряхивают в течение 10 мин для насыщения кислородом воздуха <1>. Если при 20 °C содержание растворенного кислорода выше 9 мг/л, что наблюдается во время цветения воды в водоеме (т.е. во время сильного развития водорослей), то жидкость перед анализом фильтруют через планктонную сеть N 20 <*>), а избыток кислорода удаляют, так как при инкубации возможно выделение газообразного кислорода и выбрасывание пробки. Для удаления избытка кислорода применяется один из следующих приемов: 1) отсасывание воздуха из бутыли, заполненной жидкостью наполовину (после того как вода примет температуру 20 °C), встряхивание склянки ускоряет выделение кислорода; 2) пропускание через воду сжатого воздуха. При наличии в речной воде взвеси БПК определяется как без отстаивания, так и в пробе, предварительно отстоенной в течение 30 мин непосредственно на месте отбора проб.
--------------------------------
<1> В случае транспортировки проб летом вода должна иметь температуру, близкую к нулю, для чего бутыль с водой помещается в экскурсионный ледник, а зимой воду предохраняют от замерзания, накладывая на бутыль пузырь с теплой водой.
Из бутыли с помощью сифона воду наливают в 5 калиброванных склянок с притертыми пробками емкостью 200 - 250 мл, предварительно дважды сполоснутые испытуемой водой; затем стеклянную трубку сифона опускают на дно склянки и после вытеснения воды (в трехкратном объеме) наполняют склянки водой доверху и немедленно закрывают стеклянной пробкой, чтобы не оставалось пузырька воздуха. Затем эту же воду наливают в колпачки от склянок и, перевернув последние вверх дном, вставляют их в колпачки, вытесняя из последних воду, чтобы пузырьки воздуха не попали в колпачки <2>.
--------------------------------
<2> При наличии взвешенных веществ содержимое бутыли перемешивают перед каждым заполнением легким вращательным движением.
В одной из склянок с исследуемой водой тотчас же определяют растворенный кислород. Ход определения см. стр. 70. Все остальные склянки помещают в нормальном положении в термостаты и хранят при 20 °C (с колебаниями +/- 1°) в течение определенного времени (2, 5, 7, 10 суток).
Время между заполнением склянок и фиксированием кислорода при определении его концентрации в нулевых пробах не должно превышать 15 мин.
Определение БПК с разбавлением испытуемой воды
13.3. При определении БПК загрязненных вод растворенного кислорода может не хватить для покрытия потребности воды в нем, в этом случае воду перед началом определения разбавляют дистиллированной водой, содержащей биогенные элементы.
Степень разбавления должна обеспечить остаточное содержание растворенного кислорода в конце инкубации не менее 3 - 4 мг/л и убыль кислорода 4 - 5 мг/л. Когда величина БПК неизвестна, необходимо делать несколько разбавлений, например: 1:1, 1:2, 1:4, потребность в  разбавлениях может иметь место для очень сильно загрязненных речных вод. В таких случаях разбавление приблизительно можно установить по окисляемости; кратность разбавления составляет 3/4 величины содержания кислорода, определенного по перманганатной окисляемости.
разбавлениях может иметь место для очень сильно загрязненных речных вод. В таких случаях разбавление приблизительно можно установить по окисляемости; кратность разбавления составляет 3/4 величины содержания кислорода, определенного по перманганатной окисляемости.
разбавлениях может иметь место для очень сильно загрязненных речных вод. В таких случаях разбавление приблизительно можно установить по окисляемости; кратность разбавления составляет 3/4 величины содержания кислорода, определенного по перманганатной окисляемости.Реактивы <1>
--------------------------------
<1> Все реактивы готовят на воде, не содержащей медь, свинец и цинк в количествах, превышающих указанные для разбавляющей воды.
13.4. Разбавляющая вода готовится из дистиллированной воды, которая не должна содержать медь и свинец более 0,01 мг/л, цинк свыше 1 мг/л (ионы этих металлов могут попасть в дистиллированную воду из материала перегонного куба). Вода не должна также содержать едкие щелочи, кислоты, активный хлор, хлорамины и другие бактерицидные вещества. Дистиллированную воду выдерживают в бутылях, закрытых марлей в течение нескольких дней, при комнатной температуре (целесообразно, чтобы температура воды была 20 °C). Сосуды, применяемые для этой воды, использовать для других целей нельзя (даже для приготовления разбавляющей воды). Перед применением разбавляющей воды в нее добавляют по 1 мл на 1 л следующие растворы:
а) фосфатный буферный раствор (запасной раствор). В 500 мл дистиллированной воды растворяют 8,5 г KH2PO4; 21,75 г K2HPO4; 33,4 г Na2HPO4·7H2O; 1,7 г NH4Cl и затем объем доводят до 1 л. Величина pH этого раствора должна быть 7,2;
б) раствор сульфата магния. В дистиллированной воде растворяют 22,5 г MgSO4·7H2O и разбавляют до 1 л;
в) раствор хлорида кальция. В дистиллированной воде растворяют 27,5 г хлорида кальция и разбавляют до 1 л;
г) раствор хлорида железа (III). В дистиллированной воде растворяют 0,25 г FeCl3·6H2O и разбавляют до 1 л.
После прибавления солей воду хорошо перемешивают, но не аэрируют.
Ход определения
13.5. Мерную колбу посредством сифона заполняют до половины разбавляющей водой, прибавляют точно отмеренное (пипеткой, мерной колбой или мерным цилиндром) количество исследуемой воды и доливают до метки разбавляющей водой. Колбу закрывают и содержимое тщательно перемешивают, переворачивая колбу 15 - 16 раз.
В связи с тем, что исследуемая вода может содержать небольшое количество бактерий, для интенсификации процесса можно сконцентрировать бактериальную биомассу на мембранном фильтре N 3, предварительно профильтровав воду через бумажный фильтр для удаления планктонных организмов. На 1 л разбавляющей воды достаточно обычно профильтровать 100 мл исследуемой воды. Сконцентрированную на фильтре биомассу смывают в стаканчик небольшим количеством воды и переливают в колбу с разбавляющей водой. Воду, обогащенную бактериями, описанным выше способом разливают в 5 калиброванных склянок (таким же способом, как при определении БПК в натуральной воде без разбавления). Одновременно наполняют разбавляющей водой 5 калиброванных склянок. Каждой пробе испытуемой воды должна соответствовать одна склянка с разбавляющей водой.
В одной из склянок с испытуемой водой и в одной из склянок с разбавляющей водой (нулевые пробы) тотчас же определяют растворенный кислород.
Все остальные склянки помещают в термостат и хранят при температуре 20 +/- 1 °C.
Через 2, 4 (или 5), 7, 10 суток от начала инкубации из термостата вынимают по одной склянке с испытуемой и с разбавляющей водой и определяют в них растворенный кислород и содержание нитритов. Нитриты определяют в воде, налитой в колпачок склянки, который снимают так же, как надевали (т.е. перевернув склянку вверх дном. Определение нитритов см. стр. 182). Если в пробе начался процесс нитрификации, что обнаруживается по появлению в воде нитритов в концентрации, превышающей 0,1 мг/л, то дальнейшее определение БПК не проводят. Если следы нитритов появляются на вторые или пятые сутки, то следующее определение производится через 4 и 7 суток соответственно. Если в лаборатории нет склянок с пришлифованными стеклянными колпачками, то для контроля за процессом нитрификации в термостат можно поставить дополнительно 8 неградуированных склянок любого размера, наполненных той же водой (испытуемой и разбавляющей), и в них определять содержание нитритов после каждого срока инкубации. Все склянки ставятся в термостате в сосуд, наполненный небольшим количеством дистиллированной воды, пробками вниз, так, чтобы горлышко склянок было погружено в воду, которая образует водяной затвор. Дистиллированную воду в сосуде обновляют при каждом определении. Если содержание азота нитритов в склянке будет больше 0,1 мг/л, определение растворенного кислорода производится с азидом натрия (см. стр. 77) либо по одному из следующих методов: 1) с сульфаминовой кислотой или с раствором мочевины (вариант А); 2) с бикарбонатом натрия (вариант Б).
Вариант А. В кислородную склянку, заполненную доверху пробой, добавляют 2 мл раствора сульфата марганца (40%), при этом наполненную пипетку погружают до самого дна кислородной склянки. Затем открывают верхний конец пипетки и ее медленно вынимают. Другой пипеткой прибавляют к пробе 2 мл раствора едкого кали (70%). Кончик пипетки при этом опускают только под уровень воды в горлышке кислородной склянки. Склянку осторожно закрывают, чтобы под пробкой не образовались пузырьки воздуха. Содержимое хорошо перемешивают (переворачиванием склянки) до образования хлопьевидного хорошо выпадающего осадка. Осадку дают отстояться и затем жидкость над осадком сифонируют, а к осадку добавляют 5 мл разбавленной серной кислоты. Полученный раствор переливают в колбу, склянку ополаскивают 3 раза небольшими порциями дистиллированной воды, которые вливают в ту же колбу. Затем в колбу добавляют 0,3 мл 40%-ного раствора сульфаминовой кислоты или 0,3 мл 40%-ного раствора мочевины и после этого 2 мл 15%-ного раствора KJ. Через 5 мин раствор титруют 0,05 н. раствором тиосульфата натрия с индикатором крахмалом.
Вариант Б. После растворения осадка гидроокиси марганца в колбу для титрования прибавляют 1 г бикарбоната натрия и оттитровывают выделившийся йод тиосульфатом натрия.
Надежным должно считаться определение БПК в тех пробах, где нитрификация только началась. Для большей достоверности получаемых результатов рекомендуется на инкубацию ставить параллельно 2 склянки; из найденных величин вычисляют среднюю.
Определение и расчет количества растворенного кислорода в пробе см. стр. 70.
Расчет.
БПКполн вычисляют по формуле

где A - начальное количество растворенного кислорода в воде, мг/л;
a - количество растворенного кислорода в воде после n суток инкубации, мг/л;
B - начальное количество растворенного кислорода в разбавляющей воде, мг/л;
b - количество растворенного кислорода в разбавляющей воде спустя n суток инкубации, мг/л;
C - количество разбавляющей воды, мл;
V - объем исследуемой воды, взятый для анализа, мл.
Определение БПК в воде, содержащей свободный хлор
13.6. Вода, содержащая свободный хлор, перед определением должна быть освобождена от активного хлора, для чего ее обрабатывают сульфитом натрия или тиосульфатом натрия. Для установления требующегося количества сульфита натрия отбирают отдельную порцию воды, подкисляют 0,02 н. соляной кислотой, прибавляют 10 мл 10%-ного раствора химически чистого йодида калия и раствор крахмала и оттитровывают выделившийся йод раствором сульфита натрия до исчезновения синего окрашивания.
Количество тиосульфата натрия, необходимое для восстановления свободного хлора, определяют также титрованием отдельной пробы. К 100 мл исследуемой воды прибавляют 10 мл 10%-ного раствора йодида калия и титруют 0,025 н. раствором тиосульфата натрия с применением раствора крахмала в качестве индикатора.
На 100 мл пробы, предназначенной для определения БПК, прибавляют раствор сульфита натрия или тиосульфита натрия в количестве, рассчитанном по результатам вышеописанного титрования, после чего пробу перемешивают.
Аппаратура
13.7. Кислородные склянки с хорошо притертой косо срезанной пробкой, емкостью 200 - 250 мл со стеклянным пришлифованным колпачком, калиброванные с точностью до 0,1 мл.
Мерные колбы и пипетки.
Сифон.
Бутыль.
Термостат на 20 °C с колебаниями не более +/- 1 °C.
Склянки и мерная посуда для разведения должны быть очень тщательно вымыты, так как даже небольшое загрязнение дает увеличение БПК.
Склянки должны быть обезжирены хромовой смесью, тщательно отмыты от хромовой смеси и высушены.
Реактивы
13.8. Реактивы те же, что для определения растворенного кислорода (см. стр. 76).
Сильфаминовая кислота, 40%-ный раствор.
Мочевина, 40%-ный раствор.
Бикарбонат натрия.
Тиосульфат натрия, 0,16 г безводного Na2S2O3 растворяют в 100 мл дистиллированной воды непосредственно перед применением. Раствор неустойчив.
Сульфит натрия: 3,5 г кристаллического сульфита натрия Na2SO3·7H2O растворяют в 1 л воды.
Ход определения
13.9. После добавления растворов сульфита натрия или тиосульфата натрия определение ведут так, как описано на стр. 81.
Расчет ведут так, как описано на стр. 84.
Общие положения
14.1. В природных водах алюминий находится только в малых концентрациях, которые, как правило, не превышают десятых долей мг/л, причем его обычно сопровождает железо. Соли алюминия в воде гидролизуются и выпадают в виде осадка гидроокиси. В сильнокислой среде алюминий присутствует в виде катионов, в сильнощелочной среде - в виде алюминат-ионов.
Содержание алюминия определяют в водах, при очистке которых применяли соли алюминия, а также при контроле хода работы станций водоподготовки. Для определения алюминия в питьевой и поверхностных водах предлагаются колориметрические способы анализа с применением алюминона, эриохромцианина P и стильбазо. Если надо определить алюминий только в растворе, пробу фильтруют непосредственно после отбора. Первоначальную или профильтрованную пробу консервируют добавлением 5 мл концентрированной соляной кислоты на 1 л воды.
Результаты анализа выражаются в миллиграммах алюминия в 1 л воды.
Колориметрическое определение с алюминоном
14.2. Аммониевая соль ауринтрикарбоновой кислоты - алюминон в водных растворах образует с ионами алюминия оранжево-красный комплекс. Интенсивность окраски пропорциональна концентрации алюминия. Образование комплексного соединения зависит от температуры, времени прохождения реакции и от концентрации водородных ионов. Коллоидный окрашенный раствор стабилизируют добавлением желатина. При отсутствии мешающих веществ можно этим методом определить алюминий в концентрациях не ниже 0,05 мг/л. Прямое определение можно производить до концентрации 1 г/л.
Мешающие влияния
14.3. Определению мешает присутствие железа, фторидов, полифосфатов, сероводорода, хлора, окрашенных веществ и мути.
Мешающие влияния железа можно устранить добавлением тиогликолевой кислоты.
Мешающее влияние фторидов, начиная с концентрации 0,5 мг/л, устраняют выпариванием досуха 50 мл пробы в платиновой чашке, добавлением к остатку 2 мл концентрированной серной кислоты и вторичным выпариванием на плитке всей жидкости (нельзя проводить выпаривание на открытом пламени). Остаток затем обрабатывают 2 - 5 мл концентрированной соляной кислоты, выпаривают досуха, растворяют в 10 мл горячей дистиллированной воды, подкисленной несколькими каплями HCl, и раствор фильтруют в мерную колбу емкостью 50 мл.
Остаток в чашке нагревают с 1 мл концентрированной соляной кислоты и фильтруют в ту же колбу. Чашку и фильтр промывают горячей дистиллированной водой. Остывший фильтрат в колбе доливают водой до метки. При таком ходе анализа устраняется также влияние полифосфатов, хлора и сульфидов.
Сульфиды мешают определению, начиная с концентрации 10 мг/л; их можно окислить кипячением пробы с 3%-ным раствором перекиси водорода. Остаточный хлор (после хлорирования воды) в количествах от 0,5 мг/л удаляют добавлением тиосульфата натрия.
Сильно окрашенные и мутные пробы необходимо перед анализом минерализовать. Пробу воды выпаривают с 2 - 3 мл концентрированной серной кислоты до появления белых паров, добавляют 2 мл концентрированной азотной кислоты и снова полностью выпаривают. Твердый остаток обрабатывают 10 мл дистиллированной воды и 1 мл концентрированной соляной кислоты. Фильтруют через фильтр для количественного анализа, собирая фильтрат в мерную колбу, и разбавляют объем фильтрата до первоначального объема пробы.
Аппаратура
14.4. Фотоэлектрический колориметр-нефелометр, светофильтр  или цилиндры Несслера емкостью 100 мл.
или цилиндры Несслера емкостью 100 мл.
или цилиндры Несслера емкостью 100 мл.Водяная баня.
Реактивы
14.5. n-Нитрофенол, 1%-ный раствор.
Соляная кислота ч.д.а., разбавленная (1:1).
Раствор аммиака ч.д.а., разбавленный (1:1).
Лимонная кислота, 10%-ный раствор.
Тиогликолевая кислота, примерно 1%-ный раствор: 1 мл тиогликолевой кислоты (80 - 95%-ной) разбавляют до 100 мл дистиллированной водой. Раствор может сохраняться пять суток.
Алюминон в буферном растворе, pH которого равно 4,0.
В порциях по 100 мл дистиллированной воды растворяют отдельно 138 г ацетата аммония ч.д.а., 126 мл концентрированной соляной кислоты ч.д.а., 0,9 г алюминона и 10 г желатина. Все растворы сливают в мерную колбу емкостью 1 л, дополняют до метки дистиллированной водой, перемешивают и на следующий день смесь фильтруют через стеклянную вату. Раствор сохраняется в течение 6 месяцев, pH раствора должно быть 3,8 - 4,0, что необходимо проверять электрометрическим способом.
Алюминиевые квасцы, стандартный раствор: а) запасной: 1,7582 г KAl(SO4)2·12H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л; 1 мл раствора содержит 0,100 мг Al; б) рабочий: 10 мл запасного раствора разбавляют дистиллированной водой до 1 л. Применяют всегда свежеприготовленный раствор. 1 мл раствора содержит 0,001 мг Al.
Калибровочная кривая
14.6. В набор колб отмеривают 0 - 2,0 - 5,0 - 10,0 - 20,0 - 30,0 - 40,0 - 50,0 мл рабочего стандартного раствора, что после дополнения каждого раствора до 50 мл дистиллированной водой соответствует содержанию алюминия: 0 - 0,040 - 0,10 до 1,0 мг/л. Полученные стандарты обрабатывают, как описано в ходе определения. Измеряют оптическую плотность, вычитают из нее оптическую плотность холостого определения и наносят на график против соответствующих концентраций алюминия в мг/л.
Ход определения
14.7. В колбу емкостью 250 мл помещают 50 мл пробы (если надо, предварительно разбавленной или упаренной, чтобы содержание алюминия в этом объеме оказалось в пределах 0,001 - 0,05 мг). Добавляют каплю раствора n-нитрофенола и изменяют pH раствора прибавлением раствора аммиака до появления желтой окраски индикатора. После этого добавляют по каплям соляную кислоту до исчезновения этой окраски. Затем последовательно приливают 1 мл раствора лимонной кислоты и 2 мл раствора тиогликолевой кислоты, после чего смесь тщательно взбалтывают. Добавляют 10 мл раствора алюминона в буферной смеси и колбу погружают на 15 мин в кипящую водяную баню. Смесь после этого охлаждают примерно до 20 °C и переносят в мерную колбу емкостью 100 мл или в цилиндр Несслера, доливают водой до метки и перемешивают. Не позже чем через 25 мин измеряют оптическую плотность или сравнивают с набором одновременно приготовленных стандартных растворов в цилиндрах Несслера. Из величины измеренной оптической плотности раствора вычитают оптическую плотность холостого определения с дистиллированной водой.
Расчет.
Содержание алюминия (x) в мг/л вычисляют по формуле

где c - концентрация алюминия, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,20 - 0,50 | 0,50 - 1,00 | 1,00 - 2,0 | 2,0 - 5,0 |
Округление, мг/л | 0,02 | 0,05 | 0,1 | 0,2 |
При определении алюминия в более низких концентрациях (после концентрирования пробы) и в более высоких концентрациях (после разбавления пробы) результаты округляют аналогично.
Колориметрическое определение с эриохромцианином P
14.8. Ионы алюминия образуют с эриохромцианином P в среде, pH которой равно 5,4, комплексное соединение, имеющее фиолетовую окраску, по интенсивности которой можно колориметрически определять алюминий. При отсутствии мешающих веществ без изменения объема пробы можно определить в ней алюминий при содержании его в пределах от 0,05 до 1,2 мг/л.
Мешающие влияния
14.9. Определению мешают железо (III), марганец и магний в больших концентрациях, так как эти ионы образуют аналогично окрашенные соединения. Соли железа (III) в малых концентрациях восстанавливаются солянокислым гидроксиламином, что включено в ход анализа.
Фосфаты, фториды и органические вещества понижают интенсивность окраски. В присутствии больших количеств фосфатов алюминий определяют другим способом. Органические вещества и фториды удаляют выпариванием с сильными кислотами, как это описано при определении алюминия с алюминоном.
Аппаратура
14.10. Фотометр, светофильтр  .
.
.Кюветы толщиной 2 см.
Реактивы
14.11. Эриохромцианин P, 0,1%-ный раствор. Раствор применяют только через три недели после его приготовления.
Гидроксиламин солянокислый, 10%-ный раствор.
Едкий натр ч.д.а., 10%-ный раствор.
Уксусная кислота ч.д.а., 10%-ный раствор.
Ацетат натрия + уксусная кислота, буферный раствор, pH 5,4: 42,6 г ацетата натрия ч.д.а. и 5 мл ледяной уксусной кислоты ч.д.а. растворяют в дистиллированной воде и доливают водой до 1 л. Значение pH раствора после пятикратного разбавления проверяется электрометрическим способом и доводится до pH = 5,4 раствором едкого натра или уксусной кислоты. По расходу едкой щелочи или кислоты на доведение до pH = 5,4 небольшой порции буферного раствора прибавляют соответствующее количество щелочи или кислоты ко всему объему буферного раствора.
Алюминиевые квасцы - стандартный раствор:
а) запасной; приготовление см. стр. 87; 1 мл раствора содержит 0,100 мг Al;
б) рабочий; приготовление см. стр. 87. Пользуются всегда свежеприготовленным раствором; 1 мл раствора содержит 0,001 мг Al.
Калибровочная кривая
14.12. В набор мерных колб емкостью 50 мл отмеряют 0 - 1,0 - 2,0 - 5,0 - 10,0 - 15,0 - 20,0 - 25,0 и 30,0 мл рабочего стандартного раствора, получая таким образом концентрации 0 - 0,04 - 0,08 - ... - 1,2 мг Al в 1 л.
Полученные стандарты обрабатываются, как описано в ходе определения, и значения оптической плотности после введения поправки на холостое определение наносят на график против соответствующих концентраций алюминия в мг/л.
Ход определения
14.13. В мерную колбу емкостью 50 мл отмеряют 25 мл пробы (если надо предварительно разбавленной или упаренной так, чтобы она содержала не более 0,03 мг Al). Прибавляют 0,5 мл раствора солянокислого гидроксиламина и 3 мл раствора эриохромцианина P. После смешения добавляют по каплям раствор едкого натра до появления фиолетовой окраски. Избыточную щелочь нейтрализуют добавлением по каплям уксусной кислоты до появления желтой окраски. После того приливают 10 мл буферного раствора и объем смеси доводят дистиллированной водой до 50 мл. Смесь взбалтывают и через 20 мин измеряют оптическую плотность. Аналогичным способом обрабатывают холостую пробу с дистиллированной водой и вычитают оптическую плотность холостого определения.
Расчет.
Содержание алюминия (x) в мг/л вычисляют по формуле
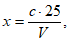
где c - концентрация алюминия, определенная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 | 1,0 - 2,0 |
Округление, мг/л | 0,01 | 0,02 | 0,05 | 0,1 |
Колориметрическое определение с помощью стильбазо
14.14. Реагент стильбазо является диаммониевой солью стильбен-4,4I-бис[(азо-1)-3,4-диоксибензол] 2,2I-дисульфокислоты и обладает структурой
 .
.Стильбазо - темно-коричневый порошок, растворы его в воде окрашены в оранжево-бурый цвет. Сильно разбавленные растворы имеют оранжево-желтую окраску, не изменяющуюся при подкислении.
Стильбазо в водных растворах образует с ионами алюминия комплексное соединение, окрашенное в розовый цвет. Интенсивность окраски пропорциональна концентрации алюминия и зависит от времени протекания реакции и величины pH раствора.
Максимальное развитие окраски происходит в интервале  . Этим методом возможно определить 0,01 мг/л Al.
. Этим методом возможно определить 0,01 мг/л Al.
. Этим методом возможно определить 0,01 мг/л Al.Мешающие влияния
14.15. Определению мешает присутствие трехвалентного железа, меди, титана, циркония, тория, бериллия.
Мешающее влияние трехвалентного железа может быть устранено восстановлением его аскорбиновой кислотой. Присутствие в воде меди до 0,05 мг также не препятствует определению алюминия после добавления аскорбиновой кислоты. Это включено в ход анализа. Титан, цирконий, торий и бериллий в поверхностных водах обычно отсутствуют. Присутствие щелочных и двухвалентных металлов не препятствует определению алюминия.
Аппаратура
14.16. Фотоэлектрический колориметр-нефелометр, светофильтр  .
.
.Пробирки с притертыми пробками, градуированные, емкостью 10 мл.
Мерные колбы емкостью 50 мл.
Микробюретки емкостью 2 мл.
Реактивы
14.17. Алюминиевые квасцы, стандартный раствор, приготовление см. стр. 87.
Рабочий, 20 мл запасного раствора разбавляют дистиллированной водой до 1 л. Применяют всегда свежеприготовленный рабочий раствор. 1 мл раствора содержит 0,002 мг Al.
Стильбазо, 0,01%-ный раствор, 0,01 г стильбазо растворяют в 100 мл дистиллированной воды. Раствор устойчив в течение двух месяцев при хранении в склянке темного стекла.
Буферный раствор, pH = 5,4 (приготовление, см. стр. 88).
Аскорбиновая кислота, свежеприготовленный 5%-ный раствор.
Подкисленная вода. К 1 л дистиллированной воды приливают 0,6 мл соляной кислоты (пл. 1,12).
Известно несколько вариантов определения алюминия с этим реагентом, ниже приведены два из них.
Вариант А. Определение алюминия
колориметрическим титрованием
14.18. В одну из двух одинаковых пробирок наливают исследуемую воду, содержащую не более 5 мкг алюминия, доводят подкисленной дистиллированной водой объем до 2 мл, прибавляют 0,15 мл свежеприготовленного 5%-ного раствора аскорбиновой кислоты, 0,5 мл 0,01%-ного раствора стильбазо и 3 мл буферного раствора, pH = 5,4. В другую пробирку вводят около 1 - 1,5 мл подкисленной воды, прибавляют такое же количество растворов аскорбиновой кислоты, стильбазо и буферного раствора. Смесь в обоих пробирках перемешивают. При наличии в исследуемой воде алюминия окраска раствора из желтой переходит в розовую.
Во вторую пробирку добавляют из микробюретки по каплям стандартный раствор алюминия, в 1 мл которого содержится 0,002 мг Al. Титрование продолжают до тех пор, пока окраска растворов в обеих пробирках не выровняется. Окраска устойчива в течение 8 ч. Если необходимо, уравнивают объем растворов прибавлением подкисленной воды. Титрование ведут со скоростью около двух капель в 1 мин в начале и одной капли в 1 мин в конце.
Расчет. Содержание алюминия (x) в мг/л находят по формуле
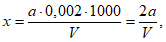
где a - количество стандартного раствора алюминия, израсходованного на титрование, мл;
V - объем пробы воды, взятой для анализа, мл.
Вариант Б. Определение алюминия
на фотоэлектрическом колориметре
14.19. В мерную колбу емкостью 50 мл отбирают такое количество исследуемой воды, чтобы содержание алюминия в пробе не превышало 15 - 17 мкг. Затем в колбу вводят 0,3 мл 5%-ного раствора аскорбиновой кислоты. Через 1,5 - 2 мин добавляют 2,5 мл буферного раствора и 5 мл раствора стильбазо. Доводят до метки дистиллированной водой и перемешивают. Спустя 5 мин измеряют оптическую плотность раствора.
В качестве холостой пробы для контроля используют дистиллированную воду, в которую введены все реактивы в той же последовательности, и через 5 мин измеряют оптическую плотность. Вводят поправку на холостую пробу и по калибровочной кривой находят содержание алюминия.
Калибровочная кривая
14.20. В ряд мерных колб емкостью 50 мл вводят 0,25; 0,5; 1,0; 2,0; 5,0; 8,0 мл рабочего раствора алюминия, которое соответствует 0,01; 0,02; 0,04; 0,08; 0,2; 0,32 мг/л Al.
В каждую из колб наливают примерно по 40 мл дистиллированной воды и последовательно вводят все реактивы, которые применяются при определении алюминия. Затем объем раствора в колбах доводят дистиллированной водой до метки и хорошо перемешивают. Спустя 5 мин производят колориметрическое определение алюминия, используя светофильтр  . Для построения калибровочной кривой откладывают на оси ординат показания прибора, а на оси абсцисс - соответствующие им величины концентраций алюминия.
. Для построения калибровочной кривой откладывают на оси ординат показания прибора, а на оси абсцисс - соответствующие им величины концентраций алюминия.
. Для построения калибровочной кривой откладывают на оси ординат показания прибора, а на оси абсцисс - соответствующие им величины концентраций алюминия.Расчет. Содержание алюминия (x) в мг/л находят по формуле

где c - концентрация алюминия, мг/л;
V - объем воды, взятой для анализа, мл.
Колориметрическое определение с 8-оксихинолином
14.21. Алюминий в кислой среде вступает в реакцию с 8-оксихинолином. Экстракт образовавшегося оксихинолята в хлороформе имеет желтую окраску, интенсивность которой пропорциональна концентрации алюминия. Экстрагированием хлороформом можно определить алюминий в широком диапазоне содержания его в пробе. При обработке 100 мл пробы 10-ю мл хлороформа можно определить содержание алюминия, начиная с концентрации 0,01 мг/л.
Мешающие влияния
14.22. Извлекаемые хлороформом и окрашивающие его органические вещества удаляют предварительной экстракцией их этим растворителем. Из мешающих определению элементов важнейший - железо. Его предварительно переводят в трехвалентную форму, окислив железо (II) персульфатом аммония, и извлекают в виде оксихинолята железа (III) хлороформом из более кислой среды  , из которой оксихинолят алюминия не извлекается. При желании железо можно попутно определить колориметрически в хлороформном экстракте. Вместе с железом (III) извлекается и медь.
, из которой оксихинолят алюминия не извлекается. При желании железо можно попутно определить колориметрически в хлороформном экстракте. Вместе с железом (III) извлекается и медь.
, из которой оксихинолят алюминия не извлекается. При желании железо можно попутно определить колориметрически в хлороформном экстракте. Вместе с железом (III) извлекается и медь.Одновременно с алюминием в хлороформный экстракт переходят висмут, индий, никель (частично), цинк (частично). Однако эти элементы обычно в водах присутствуют в количествах настолько малых по сравнению с содержанием в них алюминия, что влиянием указанных элементов в большинстве случаев можно пренебречь.
Аппаратура
14.23. Фотометр, светофильтр  .
.
.Делительные воронки, емкостью 250 - 300 мл.
Кюветы, толщиной 2 - 5 см.
Реактивы
14.24. Соляная кислота, примерно 1 н. раствор: 85 мл концентрированной соляной кислоты ч.д.а. разбавляют дистиллированной водой до 1 л.
8-оксихинолин, 2%-ный раствор в хлороформе: 2 г 8-оксихинолина ч.д.а. растворяют в хлороформе и доливают хлороформом до 100 мл.
Уксусная кислота + ацетат натрия, буферный раствор, pH = 4,5; 60 г ледяной уксусной кислоты ч.д.а. смешивают с дистиллированной водой и разбавляют до 1 л (раствор I). 82 г безводного ацетата натрия или 136 г CH3COONa·3H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л (раствор II). Смешивают 102 мл раствора I с 98 мл раствора II и доливают дистиллированной водой до 1 л.
Персульфат аммония ч.д.а. твердый.
Алюминиевые квасцы, стандартный раствор:
а) запасной: 1,7582 г KAl(SO4)2·12H2O ч.д.а. растворяют в дистиллированной воде и доводят до 1 л; 1 мл раствора содержит 0,10 мг алюминия;
б) рабочий: 100 мл запасного раствора разбавляют водой до 1 л. Применяют всегда свежеприготовленный раствор. 1 мл раствора содержит 0,010 мг алюминия.
Калибровочная кривая
14.25. Калибровочную кривую строят с учетом конечного объема экстракта и применяемых кювет. Например, для кювет длиной 3 см и конечного объема экстракта 10 мл приготавливают растворы с содержанием алюминия от 0,002 до 0,2 мг/л. В ряд делительных воронок последовательно отмеривают 0 - 0,2 - 1,0 - 5,0 - 10,0 - 15,0 - 20,0 мг рабочего раствора. Дополняют каждый раствор дистиллированной водой приблизительно до 50 мл. Таким образом получают растворы, содержащие 0,0 - 0,002 - 0,01 - 0,05 - 0,1 - 0,15 и 0,20 мг Al. После этого добавляют 2,5 - 3 мл хлороформного раствора 8-оксихинолина на 10 мл буферного раствора и экстрагируют описанным выше способом. Одновременно ставят холостой опыт с дистиллированной водой. Измеренные значения оптической плотности после вычитания из них оптической плотности холостого опыта наносятся на график против соответствующих содержаний алюминия.
Ход определения
14.26. В делительную воронку помещают 50 - 100 мл пробы, содержащей 0,005 - 0,5 мг/л алюминия. Добавляют 0,1 - 0,2 г персульфата аммония и перемешивают до растворения. Оставляют несколько минут стоять и нейтрализуют 0,1 н. раствором кислоты или щелочи. Необходимое количество кислоты или щелочи устанавливается титрованием отдельной порции воды по метиловому оранжевому. Смесь в делительной воронке подкисляют добавлением 0,8 мл 1 н. соляной кислоты на каждые 50 мл пробы (после подкисления раствор должен иметь  ). Приливают 1,5 - 2 мл раствора 8-оксихинолина в хлороформе и тщательно взбалтывают. После отделения слоев нижний слой хлороформа, окрашенный оксихинолятом железа, спускают. Добавление хлороформового раствора, экстрагирование и отделение нижнего слоя повторяется до тех пор, пока не получится бесцветный хлороформовый экстракт. Как правило, при содержании железа до 8 мг/л хватает двух экстракций.
). Приливают 1,5 - 2 мл раствора 8-оксихинолина в хлороформе и тщательно взбалтывают. После отделения слоев нижний слой хлороформа, окрашенный оксихинолятом железа, спускают. Добавление хлороформового раствора, экстрагирование и отделение нижнего слоя повторяется до тех пор, пока не получится бесцветный хлороформовый экстракт. Как правило, при содержании железа до 8 мг/л хватает двух экстракций.
). Приливают 1,5 - 2 мл раствора 8-оксихинолина в хлороформе и тщательно взбалтывают. После отделения слоев нижний слой хлороформа, окрашенный оксихинолятом железа, спускают. Добавление хлороформового раствора, экстрагирование и отделение нижнего слоя повторяется до тех пор, пока не получится бесцветный хлороформовый экстракт. Как правило, при содержании железа до 8 мг/л хватает двух экстракций.К пробе, освобожденной от железа, в делительной воронке прибавляют 2,5 - 3 мл раствора 8-оксихинолята в хлороформе и на каждые 50 мл пробы приливают по 10 мл буферного раствора. Смесь тщательно взбалтывают и оставляют стоять 1 - 2 мин. После разделения слоев нижний окрашенный в желтый цвет хлороформный экстракт собирают в мерную колбу подходящей емкости (10 - 20 мл по размеру применяемых кювет). Экстрагирование повторяют два раза, приливая каждый раз по 2,5 - 3 мл раствора оксихинолина. Экстракт в колбе дополняют хлороформом до известного объема, переносят в кювету и измеряют оптическую плотность. Из результата измерения вычитают оптическую плотность экстракта, полученного при одновременном проведении холостого опыта с дистиллированной водой.
Расчет.
Содержание алюминия (x) в м/л вычисляют по формуле

где a - содержание алюминия по калибровочной кривой, мг;
V1 - объем хлороформного экстракта из пробы, мл;
V2 - объем хлороформного экстракта при построении калибровочной кривой, мл;
V - первоначальный объем пробы, мл.
Округление результатов
Диапазон, мг/л | 0,1 - 0,2 | 0,2 - 0,5 | 0,5 - 1,00 | 1,0 - 5,0 |
Округление, мг/л | 0,01 | 0,02 | 0,05 | 0,1 |
Общие положения
15.1. В природных водах содержание железа может колебаться от тысячных долей до десятков миллиграммов в 1 л.
Раздельное определение железа (II) и (III) в воде должно выполняться непосредственно около источника водоснабжения, так как железо (II) на воздухе быстро окисляется и выпадает в осадок.
В тех случаях, когда нет возможности выполнять определения на месте отбора проб, их следует консервировать. Для этого пробы подкисляют раствором HCl (1:1) из расчета 10 мл раствора соляной кислоты на 250 мл отобранной воды.
Консервированные пробы можно хранить в холодильнике до 78 ч.
Для определения железа предлагаются колориметрические способы анализа с применением ортофенантролина, сульфосалицилата натрия и роданида.
Колориметрическое определение железа с ортофенантролином
15.2. При реакции ортофенантролина с ионами двухвалентного железа в области pH от 3 до 9 образуется красно-фиолетовое комплексное соединение. Интенсивность окраски раствора пропорциональна концентрации железа. Прямое определение возможно при содержании железа от 0,05 до 2,0 мг в 1 л. Железо (III) надо предварительно восстановить до железа (II). Для этого применяют солянокислый гидроксиламин.
Мешающие влияния
15.3. Определению мешает марганец. При его присутствии надо применять другой метод анализа. Медь мешает в концентрации, превышающей 10 мг/л. Влияние ее можно уменьшить, работая в области pH от 2,5 до 4,0.
Мешающие влияния высокого содержания органических веществ и прочих комплексных соединений железа устраняются предварительной обработкой пробы.
Для этого 50 мл пробы выпаривают с 1 мл концентрированной серной кислоты и 1 мл концентрированной азотной кислоты до появления густых паров серного ангидрида. Пробы разбавляют, доводя дистиллированной водой приблизительно до 50 мл, прибавляют 2,5 мл - примерно 0,1 н. раствора перманганата калия до появления розового окрашивания.
Горячий раствор обесцвечивают добавлением щавелевой кислоты.
Если окисленный раствор мутен, то его по охлаждении фильтруют, фильтрат доводят до 50 мл и продолжают определение описанным ниже методом.
Аппаратура
15.4. Электрофотоколориметр, светофильтр -  .
.
.Реактивы
15.5. Гидроксиламин солянокислый, 10%-ный раствор.
Ацетат натрия, 25%-ный (2 М) раствор.
ИС МЕГАНОРМ: примечание. Текст дан в соответствии с официальным текстом документа. |
1.10 - Фенантролин солянокислый, 0,5%-ный раствор.
Хранят в темной бутыли на холоде.
Стандартный раствор железа:
а) запасной - растворяют 0,8634 г железоаммонийных квасцов Fe(NH4)·(SO4)2·12H2O в мерной литровой колбе в небольшом количестве дистиллированной воды, добавляют 10 мл крепкой серной кислоты и доводят до 1 л. 1 мл раствора содержит 0,1 мг Fe;
б) рабочий - 50,0 мл запасного стандартного раствора разбавляют до 1 л дистиллированной водой, каждый раз приготовляют свежий раствор; 1 мл раствора содержит 0,005 мг железа.
Калибровочная кривая
15.6. В мерные колбы емкостью 50 мл отмеривают от 0 до 20 мл рабочего стандартного раствора железа и доводят объемы дистиллированной водой до метки. В приготовленной таким путем серии растворов, содержащих 0 - 0,05 - 0,10 ... - 2,0 мг/л железа, определяют железо указанным выше способом. Вычитают оптическую плотность холостой пробы. Полученные значения наносят на график против соответствующих концентраций железа в мг/л.
Ход определения
15.7. В мерную колбу емкостью 50 мл помещают такой объем анализируемой пробы, чтобы в нем содержалось 0,0025 - 0,1 мг железа, слегка подкисляют соляной кислотой, если она имеет нейтральную или щелочную реакцию. Затем, пользуясь другой аликвотной порцией, находят необходимое количество ацетата натрия для нейтрализации пробы (pH = 3,5) путем титрования в присутствии индикатора бромфенолового синего или метилового оранжевого и требуемое количество ацетата вводят в мерную колбу.
Прибавляют 1 мл раствора солянокислого гидроксиламина, 1 мл раствора ортофенантролина, дают постоять 10 - 15 мин, разбавляют дистиллированной водой до 50 мл и перемешивают.
Измеряют оптическую плотность раствора, из которой вычитают оптическую плотность холостой пробы, приготовленной таким же способом, но с дистиллированной водой вместо пробы. Результаты определений находят по калибровочной кривой.
Этим же способом можно определить содержание железа (II) в присутствии железа (III), но тогда опускают восстановление железа (III) гидроксиламином.
Расчет. Содержание железа (x) в мг/л вычисляют по формуле

где c - концентрация железа, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл.
Колориметрическое определение с сульфосалицилатом натрия
15.8. Метод основан на том, что сульфосалициловая кислота (или ее натриевая соль) образует с солями железа окрашенные комплексные соединения, причем в слабокислой среде сульфосалициловая кислота реагирует только с солями железа (III) (красное окрашивание), а в слабощелочной среде - как с солями железа (III), так и с солями железа (II) (желтое окрашивание).
Аппаратура
15.9. Фотометр, светофильтр 400 - 413 нм.
Кюветы длиной 5 см.
Реактивы
15.10. Сульфосалициловая кислота, 10%-ный раствор, или сульфосалицилат натрия, насыщенный раствор.
Аммиак, разбавленный раствор. Смешивают 200 мл концентрированного раствора аммиака с 300 мл воды.
Стандартный раствор железа: приготовление см. в описании колориметрического метода определения железа с ортофенантролином; 1 мл раствора содержит 0,1 мг железа.
Соляная кислота, разбавленный (3:2) раствор.
Мешающее влияние высокого содержания органических веществ устраняется как указано на стр. 95.
Ход определения
15.11. В колбу емкостью 100 - 150 мл наливают пипеткой 50 мл анализируемой воды, затем прибавляют 5 мл раствора сульфосалицилата натрия или сульфосалициловой кислоты, 5 мл раствора аммиака и перемешивают. Спустя 10 мин интенсивность окрашенного раствора измеряют в фотоколориметре с синим светофильтром  . Результаты определения находят по калибровочной кривой. Чувствительность метода при цветности воды до 10° - 0,05 мг/л Fe. С увеличением цветности природных вод до 100° чувствительность метода снижается до 0,2 мг/л Fe.
. Результаты определения находят по калибровочной кривой. Чувствительность метода при цветности воды до 10° - 0,05 мг/л Fe. С увеличением цветности природных вод до 100° чувствительность метода снижается до 0,2 мг/л Fe.
. Результаты определения находят по калибровочной кривой. Чувствительность метода при цветности воды до 10° - 0,05 мг/л Fe. С увеличением цветности природных вод до 100° чувствительность метода снижается до 0,2 мг/л Fe.Расчет. Содержание железа рассчитывают по формуле, приведенной в методе определения железа с ортофенантролином (стр. 96).
Колориметрическое определение с роданидом
15.12. Метод основан на взаимодействии в сильно кислой среде ионов железа (III) с ионами роданида. В зависимости от концентрации ионов роданида образуется ряд комплексных ионов кроваво-красного цвета, обуславливающих различную интенсивность окрашенного раствора:

где n - число ионов роданида, связанных в железороданидный комплекс; n - может изменяться от 1 до 6.
Для колориметрирования необходимы строго определенные соотношения в растворе между концентрацией различных комплексных ионов. Это достигается добавлением реактива в количестве, дающем возможность создавать постоянный избыток ионов роданида.
Для определения общего содержания железа железо (II) предварительно окисляют персульфатом аммония или перекисью водорода до железа (III). Определению мешает ряд металлов, как, например, медь, висмут и кобальт.
Мешающее влияние высокого содержания органических веществ и трудно разлагаемых комплексов железа устраняется выпариванием пробы с азотной и серной кислотами, как описано в колориметрическом методе определения железа с ортофенантролином (стр. 94).
Аппаратура
15.13. Фотоэлектрический колориметр, ФЭК-Н-57, синий светофильтр N 3  . В случае отсутствия фотоколориметра колориметрирование может выполняться в одинаковых фарфоровых чашках емкостью 25 или 50 мл со стеклянными палочками для перемешивания.
. В случае отсутствия фотоколориметра колориметрирование может выполняться в одинаковых фарфоровых чашках емкостью 25 или 50 мл со стеклянными палочками для перемешивания.
. В случае отсутствия фотоколориметра колориметрирование может выполняться в одинаковых фарфоровых чашках емкостью 25 или 50 мл со стеклянными палочками для перемешивания.Реактивы
15.14. Железоаммонийные квасцы, стандартный раствор, приготовление см. стр. 95. 1 мл раствора содержит 0,1 мг железа.
Роданид аммония или калия, 50%-ный раствор.
Персульфат аммония или калия.
Соляная кислота, свободная от железа разбавленная (1:1).
Ход определения
15.15. При общем содержании железа в воде до 3 мг/л определение железа выполняется без разбавления пробы дистиллированной водой.
В мерную колбу емкостью 50 мл наливают до метки испытуемую воду, добавляют 1 мл раствора соляной кислоты (1:1) несколько кристаллов персульфата аммония, после 3-минутного перемешивания вводят 1 мл 50%-ного раствора роданида аммония, снова перемешивают и спустя 3 мин колориметрируют.
Если раствор соляной кислоты добавлялся ранее для консервирования пробы, дополнительного подкисления в момент определения не требуется. По калибровочному графику находят общее содержание железа в испытуемой воде.
При визуальном колориметрировании в фарфоровых чашках в левую чашку наливают 25 мл испытуемой воды, в правую - такое же количество дистиллята.
После добавления в обе чашки реактивов раствор в правой чашке должен оставаться бесцветным. В случае появления окраски в правой чашке определение повторяют, тщательно вымыв посуду и проверив реактивы.
После добавления реактивов в правую чашку по каплям приливают стандартный раствор железа, одновременно перемешивая содержимое до тех пор, пока интенсивность окраски в обеих чашках не станет одинаковой.
При общем содержании железа до 0,4 мг/л следует пользоваться стандартным раствором, в котором в 1 мл содержится 0,01 мг железа. При большем содержании железа - раствором, в 1 мл которого содержится 0,1 мг железа.
Расчет. При визуальном методе определения железа его содержание (x) в мг/л вычисляют по формуле
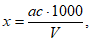
где a - количество мл стандартного раствора, пошедшее на титрование;
c = 0,1 - если использовался стандартный раствор, в 1 мл которого содержится 0,1 мг железа; c = 0,01 - если в 1 мл содержится 0,01 мг железа;
V - объем воды, взятый на определение, мл.
Определение трехвалентного железа производят так же, как и определение общего содержания железа, за исключением того, что не добавляют персульфат аммония для окисления двухвалентного железа в трехвалентное.
Определение содержания трехвалентного железа
сульфосалициловым методом
15.16. Все определения производят так же, как и определение общего содержания железа, за исключением того, что вместо аммиака в определяемую пробу воды вливают 0,1 мл соляной кислоты. Спустя 10 мин возникшую окраску анализируемого раствора определяют с помощью фотоколориметра. При измерении интенсивности окраски применяют светофильтры  .
.
.Результаты определения рассчитывают по той же формуле, что и при определении содержания общего железа. Чувствительность метода при цветности воды до 15° - 0,05 мг/л Fe.
16. ИОНЫ АММОНИЯ И АММИАК <1>
--------------------------------
<1> В дальнейшем суммарное содержание ионов аммония и свободного аммиака будет выражаться словам "аммиак".
Общие положения
16.1. Ионы аммония и аммиака могут содержаться в подземных водах в результате жизнедеятельности микроорганизмов или в результате загрязнения водоносного горизонта сточными водами. В поверхностных водах аммиак содержится в небольших количествах в период вегетации в результате разложения белковых веществ или при их загрязнении стоками. Вследствие жизнедеятельности нитрифицирующих бактерий содержание аммиака в водоемах снижается при одновременном образовании нитратов.
Отношение концентраций свободного аммиака (NH3) и ионов аммония 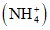 зависит от концентрации водородных ионов (см. табл. 9).
зависит от концентрации водородных ионов (см. табл. 9).
зависит от концентрации водородных ионов (см. табл. 9).Таблица 9
от pH воды
pH | 6 | 7 | 8 | 9 | 10 | 11 |
NH3, % | 0 | 1 | 4 | 25 | 78 | 96 |
100 | 99 | 96 | 75 | 22 | 4 |
Для определения аммиака приводятся два метода: 1) непосредственного колориметрического определения с реактивом Несслера в подземных и чистых поверхностных водах; 2) отгонка с колориметрическим или объемным окончанием (соответственно концентрации аммиака в пробе) для определения в загрязненных поверхностных водах. Если определение аммиака производится не сразу же после отбора проб, то их консервируют прибавлением 1 мл концентрированной серной кислоты или 2 - 4 мл хлороформа на 1 л пробы.
Качественное определение
16.2. К 10 мл пробы прибавляют несколько кристалликов сегнетовой соли и 0,5 мл реактива Несслера. Желтое окрашивание раствора, помутнение или желто-коричневый осадок указывают на присутствие аммиака. В присутствии повышенного количества органических веществ или гуминовых веществ, которые вызывают усиление коричневой окраски после подщелачивания, необходимо провести холостой опыт, добавив сегнетовую соль и 0,5 мл 15%-ного раствора едкого натра на 10 мл пробы.
Непосредственное колориметрическое определение
с реактивом Несслера
16.3. Аммиак реагирует в щелочной среде с йодомеркуриатом калия с образованием осадка желто-коричневого цвета. При низкой концентрации аммиака образуется коллоидный раствор, пригодный для колориметрического определения.
Низший предел определения равен 0,05 мг  в 1 л. Без разбавления можно определять не более 4 мг
в 1 л. Без разбавления можно определять не более 4 мг  в 1 л.
в 1 л.
Мешающие влияния
16.4. Определению мешают амины, хлорамины, ацетон, альдегиды, спирты и некоторые органические соединения, реагирующие с реактивом Несслера. В их присутствии применяют определение аммиака с отгонкой.
Определению также мешают компоненты жесткости воды, железо, сульфиды, хлор, мутность.
Мешающее влияние жесткости воды устраняют прибавлением раствора сегнетовой соли или комплексона III.
Большое количество железа, сульфиды и муть удаляются осветлением воды солью цинка. К 100 мл пробы прибавляют 1 мл раствора сульфата цинка (100 г ZnSO4·7H2O ч.д.а. растворяют в дважды дистиллированной воде и разбавляют до 1 л) и смесь тщательно перемешивают. Затем pH смеси доводят до 10,5 добавлением раствора 25%-ного едкого кали или едкого натра. После взбалтывания и хлопьеобразования осадок отделяют центрифугированием или фильтрованием через стеклянный фильтр. Увеличение объема жидкости необходимо учесть при расчете.
Мешающее влияние хлора устраняют добавлением раствора тиосульфата (3,5 г Na2S2O3 ч.д.а. растворяют в дважды дистиллированной воде и доводят до 1 л). Для удаления 0,5 мг хлора достаточно прибавить 1 мл этого раствора.
Аппаратура
16.5. Фотометр, фиолетовый светофильтр  .
.
.Кюветы, длиной 1 - 5 см или набор цилиндров Несслера емкостью 50 мл.
Мерные колбы емкостью 50 мл.
Реактивы
16.6. Дважды дистиллированная безаммиачная вода. В дважды дистиллированной воде устраняют следы аммиака фильтрованием через катионит в H+-форме.
Реактив Несслера. Растворяют 100 г HgJ2 ч.д.а. и 70 г KJ ч.д.а. в небольшом количестве дважды дистиллированной воды и смешивают с раствором едкого натра, приготовленным растворением 160 г NaOH ч.д.а. в 500 мл дистиллированной воды. Смесь доводят дважды дистиллированной водой до 1 л. Применяется прозрачный раствор после отстаивания в течение 4 ч.
Тартрат натрия и калия (сегнетовая соль), 50%-ный раствор: 50 г KNaC4O6·4H2O ч.д.а. растворяют в дважды дистиллированной воде, разбавляют до 100 мл и прибавляют 0,2 - 0,5 мл реактива Несслера. Раствор можно применять после его осветления.
Комплексон III, 50%-ный раствор: 10 г NaOH ч.д.а. растворяют в 60 мл дважды дистиллированной воды и в полученном растворе растворяют 50 г комплексона III. Смесь доводят дважды дистиллированной водой до 100 мл.
Едкий натр ч.д.а., 15%-ный раствор в дважды дистиллированной воде.
Хлорид аммония, стандартный раствор:
а) запасной, 0,2965 г NH4Cl ч.д.а. растворяют в дважды дистиллированной воде и разбавляют до 1 л; 1 мл раствора содержит 0,100 мг  ; раствор должен быть всегда свежеприготовленным;
; раствор должен быть всегда свежеприготовленным;
б) рабочий, 50,0 мл запасного раствора доводят дважды дистиллированной водой до 1 л; раствор должен быть всегда свежеприготовленным; 1 мл раствора содержит 0,005 мг  .
.
Реактивы должны быть без аммиака, что контролируется холостым опытом.
Калибровочная кривая
16.7. В мерные колбы или в цилиндры Несслера последовательно прибавляют 0 - 0,5 - 1,0 - 2,0 - 4,0 - 6,0 - 8,0 - 10,0 - до 40 мл рабочего стандартного раствора хлорида аммония и объем доводят дважды дистиллированной водой до 50 мл. Растворы с концентрациями 0 - 0,20 - 0,40 - 0,60 - 0,80 - 1,0 - до 4,0 мг 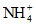 в 1 л обрабатывают, как описано в ходе определения. Определяют оптическую плотность, вычитают оптическую плотность холостой пробы и строят калибровочную кривую зависимости оптической плотности от концентрации ионов аммония, в мг/л. (При сравнении в цилиндрах Несслера приготовляют шкалу только до 2 мг
в 1 л обрабатывают, как описано в ходе определения. Определяют оптическую плотность, вычитают оптическую плотность холостой пробы и строят калибровочную кривую зависимости оптической плотности от концентрации ионов аммония, в мг/л. (При сравнении в цилиндрах Несслера приготовляют шкалу только до 2 мг  в 1 л).
в 1 л).
Ход определения
16.8. К 50 мл первоначальной пробы или к 50 мл осветленной пробы или к меньшему объему, доведенному до 50 мл дважды дистиллированной водой, прибавляют 1 - 2 капли раствора комплексона III или сегнетовой соли и смесь тщательно перемешивают. Для очень жестких вод нужно прибавить 0,5 - 1 мл раствора сегнетовой соли или комплексона III. Затем прибавляют 1 мл реактива Несслера и снова перемешивают. Через 10 мин колориметрируют или сравнивают с серией стандартов, приготовленных в цилиндрах Несслера. Окраска смеси не меняется в течение 30 мин. Из величины оптической плотности вычитают ее значение для холостого опыта. Если нужно, вычитают оптическую плотность пробы, к которой вместо реактива Несслера прибавляют 1 мл 15%-ного раствора едкого натра.
Ставят холостой опыт с дважды дистиллированной водой и вычитают значение плотности из величины ее, полученной для пробы.
Расчет. Содержание ионов аммония  в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где c - найденная концентрация аммиака, мг/л;
V - объем пробы, взятой для определения, мл;
0,0554 - коэффициент пересчета.
Округление результатов
Диапазон, мг/л | 0,05 - 2,00 | 2,0 - 5,0 | 5,0 - 10,0 | 10,0 - 20,0 и т.д. |
Округление: | ||||
мг/экв | 0,002 | 0,005 | 0,01 | 0,02 |
мг | 0,05 | 0,1 | 0,2 | 0,5 |
Определение с перегонкой
16.9. Определение заключается в выделении аммиака перегонкой из щелочной среды и определении его в дистилляте колориметрическим или объемным методом. Для выделения аммиака можно применять обыкновенную перегонку или перегонку с водяным паром. Пробу подщелачивают фосфатным буферным раствором до получения смеси pH = 7,4.
В зависимости от содержания в пробе аммиака для его поглощения в приемнике применяют серную кислоту различной нормальности или борную кислоту. Количество серной кислоты, пошедшее на нейтрализацию перегнанного аммиака, определяют обратным титрованием раствором едкого натра. При применении борной кислоты перегнанный аммиак определяют прямым титрованием раствором серной кислоты. Титруют по метиловому красному или со смешанным индикатором, состоящим из метилового красного и метиленового синего.
Колориметрическое определение аммиака в дистилляте с реактивом Несслера можно применять в том случае, если концентрация аммиака в 100 мл дистиллята меньше 0,1 мг. При более высокой концентрации аммиака применяется объемный метод. Титруют 0,02 н., 0,1 н. и 1 н. растворами кислот при содержании аммиака в 100 мл дистиллята, соответственно равном 5, 25 и выше 25 мг.
Мешающие влияния
16.10. Определению мешает свободный хлор, который перед перегонкой устраняют прибавлением тиосульфата натрия.
Кальций при концентрациях выше 250 мг/л влияет на установление pH. В этом случае раствор подщелачивают добавлением 40 мл буферного фосфатного раствора и смесь обрабатывают кислотой или щелочью для получения pH = 7,4.
Летучие органические соединения, которые мешают колориметрическому определению аммиака в дистилляте, устраняют кипячением слабо подкисленной пробы.
Аппаратура
16.11. Перегонный аппарат для простой перегонки или аппарат для перегонки с водяным паром.
Реактивы
16.12. Дважды дистиллированная, свободная от аммиака вода.
Буферный раствор, pH = 7,4: растворяют 14,3 г KH2PO4 (безводного) ч.д.а. и 68,8 г K2HPO4 (безводного) ч.д.а. в дважды дистиллированной воде и разбавляют до 1 л. Значение pH = 7,4 при необходимости корректируют добавлением едкого кали или соляной кислоты.
Серная кислота, приблизительно 1 н. раствор: 28 мл H2SO4 ч.д.а. (концентрированной) отмеривают и прибавляют к 500 мл дважды дистиллированной воды и после перемешивания разбавляют до 1 л.
Серная кислота, приблизительно 0,1 н. раствор: отмеривают 2,8 мл концентрированной H2SO4 ч.д.а. и добавляют к 500 мл дважды дистиллированной воды, перемешивают и разбавляют до 1 л.
Серная кислота, 0,02 н. раствор: 200 мл 0,1 н. H2SO4 разбавляют дважды дистиллированной водой и доводят до 1 л. Титр или поправку к этому раствору проверяют титрованием 0,02 н. раствором NaOH.
Едкий натр, 1 н. раствор: 40 г NaOH ч.д.а. растворяют в дважды дистиллированной воде и разбавляют до 1 л. Титр или поправку определяют титрованием 1 н. раствором щавелевой кислоты.
Едкий натр, 0,1 н. раствор: 4 г NaOH ч.д.а. растворяют в холодной свежепрокипяченной, дважды дистиллированной воде и разбавляют до 1 л. Титр или поправку определяют титрованием раствором щавелевой или другой кислоты той же нормальности.
Едкий натр, 0,02 н. раствор: 200 мл 0,1 н. раствора едкого натра разбавляют холодной, свежепрокипяченной, дважды дистиллированной водой до 1 л. Определяют титр или поправку так же, как у 0,1 н. раствора NaOH. Титруют 0,02 н. раствором щавелевой кислоты, который приготовляют разбавлением 200 мл 0,1 н. раствора щавелевой кислоты, свежеприготовленной и охлажденной дистиллированной водой при 20 °C до 1 л.
Борная кислота, 2%-ный раствор: 20 г H3BO3 ч.д.а. растворяют в дважды дистиллированной воде и разбавляют до 1 л.
Метиловый красный, 0,1%-ный раствор, индикатор: 0,1 г натриевой соли метилового красного растворяют в 100 мл 96%-ного спирта.
Метиловый красный и метиленовая синяя, смешанный индикатор: 2 объема 0,2%-ного раствора метилового красного в 96%-ном спирте смешивают с 1 объемом 0,2%-ного раствора метиленовой синей в 96%-ном спирте.
Реактив Несслера (приготовление см. стр. 100).
Ход определения
16.13. В перегонную колбу отмеривают 100 мл (или меньше или больше, от 10 до 500 мл, в зависимости от ожидаемого содержания аммиака) пробы, которую при необходимости разбавляют дважды дистиллированной водой приблизительно до 250 мл. Если необходимо, смесь нейтрализуют 1 н. раствором едкого натра до pH = 7 (определяется индикаторной бумажкой). Пробы, которые подщелачивались, нейтрализовать не надо.
В приемник, который представляет собой колбу для титрования емкостью 500 мл, наливают 25 мл серной кислоты соответствующей нормальности или 25 мл 2%-ного раствора борной кислоты и, добавляя дважды дистиллированную воду, устанавливают объем жидкости так, чтобы конец холодильника был в нее погружен.
В перегонную колбу прибавляют 100 мл фосфатного буферного раствора. Значение pH смеси в колбе не должно отличаться более чем на +/- 0,2 от pH = 7,4.
В приемник отгоняют около 200 - 100 мл жидкости. При применении аппарата для перегонки с паром в приемник перегоняют с водяным паром по меньшей мере 50 мл дистиллята. После этого приемник опускают ниже и собирают по каплям еще около 5 мл дистиллята. Затем прерывают перегонку и конец холодильника ополаскивают дважды дистиллированной водой.
Аммиак, перегнанный в приемник, определяют либо объемным методом, либо колориметрически.
При определении объемным методом с применением серной кислоты содержимое приемника титруют раствором едкого натра соответствующей концентрации по метиловому красному или со смешанным индикатором. Титровать надо раствором той же нормальности, какой был применен для поглощения аммиака. Параллельно определяют количество раствора щелочи, израсходованного на титрование 25,0 мл кислоты, помещенной в приемник. Титрование ведут до получения такой же окраски индикатора.
При определении объемным методом с применением борной кислоты проводят холостой опыт; 25,0 мл раствора борной кислоты титруют 0,02 н. раствором серной кислоты по метиловому красному или со смешанным индикатором. Дистиллят титруют тем же раствором до такой же окраски, какая была получена при титровании холостого опыта.
При колориметрическом определении содержимое приемника обрабатывают 1 н. кислотой до достижения pH = 6, переливают в мерную колбу емкостью 250 мл и объем доводят до метки дважды дистиллированной водой. В 50 мл аликвотной части определяют аммиак способом, указанным на стр. 102.
Расчет. Содержание ионов аммония  в мг/л (x) или в мг-экв/л (y) вычисляют следующим образом. При определении объемным методом с поглощением серной кислотой:
в мг/л (x) или в мг-экв/л (y) вычисляют следующим образом. При определении объемным методом с поглощением серной кислотой:
в мг/л (x) или в мг-экв/л (y) вычисляют следующим образом. При определении объемным методом с поглощением серной кислотой:

где a - количество раствора едкого натра, пошедшего на титрование 25,0 мл раствора серной кислоты, мл;
b - количество раствора едкого натра, израсходованного при титровании дистиллята, мл;
k - поправка титрованного раствора едкого натра;
N - нормальность титрованного раствора едкого натра;
V - объем пробы, взятой для определения, мл.
При использовании 2%-ного раствора борной кислоты:
a - количество 0,02 н. раствора серной кислоты, пошедшего на титрование дистиллята, мл;
b - количество 0,02 н. раствора серной кислоты, пошедшего на титрование в холостом опыте, мл;
k - поправка титрованного раствора серной кислоты;
N - нормальность титрованного раствора серной кислоты.
При колориметрическом определении расчет ведут по формулам:


где c - концентрация аммиака, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения при перегонке, мл.
Округление результатов. Результаты, полученные при объемном определении, округляют до 0,1 мг в области до 10 мг/л, до целых миллиграммов в области до 100 мг/л и т.д. Результаты колориметрического определения округляют так же, как при непосредственном колориметрическом определении, с реактивом Несслера.
Общие положения
17.1. Кадмий в обычных водах, как правило, отсутствует. Встречается он иногда в шахтных водах, сточных водах некоторых производств. Кадмий может присутствовать в ионной форме в виде гидроокиси или карбоната. В сточных водах и поверхностных водах, загрязненных стоками, кадмий может быть также в виде комплексного цианида или тартрата.
Для определения кадмия во всех типах вод приводится экстрактивный колориметрический метод с дитизоном; этот метод можно применять для определения кадмия в концентрациях от сотых до целых миллиграммов в 1 л.
В нейтральной и щелочной средах кадмий адсорбируется на стенках сосудов. Пробу необходимо поэтому консервировать добавлением 0,5 мл концентрированной азотной кислоты на каждые 100 мл пробы. Пробы, содержащие цианиды, не консервируют. Результаты определений выражаются в миллиграммах кадмия на 1 л воды.
Колориметрическое определение с дитизоном
17.2. Ионы двухвалентного кадмия экстрагируют раствором дитизона в четыреххлористом углероде из сильнощелочных растворов, содержащих тартрат-ионы. Интенсивность окраски экстракта, окрашенного в красный цвет дитизоната кадмия, пропорциональна концентрации кадмия. Приведенным способом при анализе 50 мл неразбавленной пробы можно определить 0,01 - 0,5 мг кадмия в 1 л воды.
Мешающие влияния
17.3. Пробы, содержащие значительные количества органических веществ, надо минерализовать способом, описанным при определении меди (см. стр. 126).
В сильнощелочной среде с дитизоном помимо кадмия реагируют серебро, медь, никель и кобальт.
Описанным способом можно определить 0,05 - 0,5 мг кадмия в присутствии 5 мг меди и кобальта и 50 мг серебра, ртути и никеля в 1 л пробы.
Большие количества серебра осаждают в виде хлорида.
Если проба содержит цианиды, то последние необходимо разрушить кипячением в течение 10 мин под хорошей тягой при добавлении 0,5 мл концентрированной серной кислоты на каждые 100 мл раствора. К 50 мл первоначальной пробы или полученной после разложения цианида прибавляют 0,2 мл концентрированной соляной кислоты. Перемешивают, оставляют на 2 ч и фильтруют. Фильтр промывают дистиллированной водой. Незначительное количество серебра, остающееся в фильтрате, извлекают ниже описанной экстракцией.
Медь, ртуть и малые количества серебра удаляют из пробы экстрагированием раствором дитизона при pH = 2. К 50 мл пробы (или раствора, полученного после минерализации пробы или удаления из нее большей части серебра) прибавляют 5 мл 20%-ного раствора тартрата натрия и калия и доводят до pH = 2 добавлением концентрированной соляной кислоты или раствора аммиака. Пробу экстрагируют в делительной воронке порциями по 5 мл 0,1%-ного раствора дитизона в хлороформе до тех пор, пока зеленая окраска дитизона в растворе не перестанет изменяться. После этого пробу промывают порциями по 10 мл хлороформа, пока хлороформный слой не останется бесцветным. Окончательно пробу промывают 2 раза встряхиванием с 5 мл четыреххлористого углерода. Если за экстракцией указанных элементов должно последовать экстрактивное удаление никеля, промывку четыреххлористым углеродом не проводят.
Никель удаляют диметилглиоксимом, причем одновременно с этим маскируется кобальт. К 50 мл первоначальной пробы или пробы из раствора, полученного после минерализации, прибавляют 10 мл 20%-ного раствора сегнетовой соли (к пробе, освобожденной от серебра, меди и ртути экстрагированием, прибавляют только 5 мл этого раствора). Приливают раствор аммиака до  , прибавляют 5 мл 1%-ного раствора диметилглиоксима в 96%-ном этиловом спирте и смесь тщательно взбалтывают 30 сек. Затем экстрагируют порциями по 10 мл хлороформа, пока не будет извлечен весь избыток диметилглиоксима, после чего окончательно промывают пробу встряхиванием с 5 мл четыреххлористого углерода.
, прибавляют 5 мл 1%-ного раствора диметилглиоксима в 96%-ном этиловом спирте и смесь тщательно взбалтывают 30 сек. Затем экстрагируют порциями по 10 мл хлороформа, пока не будет извлечен весь избыток диметилглиоксима, после чего окончательно промывают пробу встряхиванием с 5 мл четыреххлористого углерода.
, прибавляют 5 мл 1%-ного раствора диметилглиоксима в 96%-ном этиловом спирте и смесь тщательно взбалтывают 30 сек. Затем экстрагируют порциями по 10 мл хлороформа, пока не будет извлечен весь избыток диметилглиоксима, после чего окончательно промывают пробу встряхиванием с 5 мл четыреххлористого углерода.Определению кадмия не мешает присутствие свинца, висмута, мышьяка, сурьмы, олова, хрома, алюминия, железа, марганца, цианидов, роданидов, фосфатов, сульфитов, тиосульфатов и других ионов, обычно присутствующих в водах в концентрациях ниже 50 мг/л.
Если присутствует цинк в концентрации ниже 50 мг/л, то определению кадмия в приведенных концентрациях он не мешает. Если соотношение цинк:кадмий превышает 500:1, кадмий экстрагируется не полностью и результат определения получается с отрицательной ошибкой.
В пробе не должны присутствовать окислители, окисляющие дитизон (появляется коричневое окрашивание продуктов окисления, мешающее определению). Такое влияние оказывают, например, хлор, йод, бром, перекиси и т.д. Из пробы эти вещества устраняют кипячением.
Экстрагирующиеся красители удаляют предварительной экстракцией пробы четыреххлористым углеродом. Экстракцию проводят порциями по 5 мл до тех пор, пока последний экстракт не окажется бесцветным.
Дальше определение проводят по приведенному выше способу.
Аппаратура
17.4. Фотометр, светофильтр  .
.
.Кюветы толщиной 1 - 5 см.
Реактивы
17.5. Дважды перегнанная дистиллированная вода. Получают ее повторной перегонкой дистиллированной воды в стеклянном приборе. Применяется для приготовления всех реактивов и для разбавления проб.
Соляная кислота ч.д.а., концентрированная.
Раствор аммиака ч.д.а., концентрированный.
Тартрат натрия и калия (сегнетова соль), 20%-ный раствор: 50 г NaKC4H4O6·4H2O ч.д.а. растворяют в дважды дистиллированной воде, дополняют такой же водой до 250 мл и в делительной воронке экстрагируют порциями по 10 мл раствора дитизона в четыреххлористом углероде до тех пор, пока зеленая окраска раствора дитизона не перестанет изменяться. Оставшийся в водном растворе дитизон и желтый продукт его окисления удаляют экстрагированием четыреххлористым углеродом, пока экстракт не получится бесцветным. Раствор сохраняют в полиэтиленовой склянке.
Едкий натр, 10%-ный раствор. Сохраняют в полиэтиленовой склянке.
Едкий натр, 2%-ный раствор. Сохраняют в полиэтиленовой склянке.
Четыреххлористый углерод, ч.д.а. или очищенный перегонкой: 500 мл четыреххлористого углерода кипятят со 100 мл 5%-ного раствора едкого натра с обратным холодильником в течение 2 ч. После этого отделяют слой органического растворителя и промывают его 100 мл дважды дистиллированной воды. Высушивают над хлоридом кальция и перегоняют с добавкой окиси кальция.
Дитизон, раствор в четыреххлористом углероде:
а) запасной: 0,1 г дитизона ч.д.а. растворяют в 500 мл четыреххлористого углерода. Раствор сохраняют на холоде в коричневой склянке;
б) рабочий: 75 мл запасного раствора дитизона экстрагируют равным объемом 1%-ного раствора аммиака в дважды дистиллированной воде. В водный слой переходит аммонийная соль дитизона, а в слое органического растворителя останутся продукты его окисления, окрашенные в желтый цвет. После разделения слоев аммиачный раствор дважды промывают четыреххлористым углеродом. После этого к аммиачному раствору прибавляют 75 мл четыреххлористого углерода, смесь подкисляют разбавленной (1:1) соляной кислотой и встряхивают. Выделенный дитизон перейдет в слой четыреххлористого углерода. После отделения водного слоя раствор дитизона промывают тщательным встряхиванием со 100 мл дважды дистиллированной воды. Отделяют водный слой, а слой четыреххлористого углерода разбавляют этим же растворителем до 150 мл. Раствор устойчив примерно одну неделю. Его сохраняют на холоду и в темной склянке.
Хлорид кадмия, стандартный раствор:
а) запасной: 0,100 г металлического кадмия ч.д.а. растворяют в 25 мл разбавленной (1:4) соляной кислоты и доливают дважды дистиллированной водой до 1 л. Сохраняют в полиэтиленовой склянке; 1 мл раствора содержит 0,100 мг кадмия;
б) рабочий: 10,0 мл запасного раствора смешивают с 5 мл концентрированной соляной кислоты и дополняют дважды дистиллированной водой до 1 л. Всегда применяют свежеприготовленный раствор. 1 мл раствора содержит 0,001 мг кадмия.
Калибровочная кривая
17.6. В делительные воронки отмеряют по 0 - 0,5 - 2,5 - 5,0 - 10,0 - 15,0 - 20,0 - 25,0 мл рабочего стандартного раствора и объем доводят до 50 мл дважды дистиллированной водой. Концентрации этим способом приготовленных стандартных растворов равны 0,00 - 0,01 - 0,05 - ... - 0,50 мг кадмия в 1 л. Растворы обрабатывают описанным выше способом. Измеренные значения оптической плотности после вычитания из них оптической плотности холостого определения наносят на график против соответствующих концентраций кадмия в мг/л.
Ход определения
17.7. В делительную воронку емкостью 150 мл отмеряют 50 мл пробы, если надо предварительно разбавленной бидистиллятом или сконцентрированной выпариванием так, чтобы в этом объеме она содержала 0,002 - 0,020 мг кадмия. Прибавляют 10 мл раствора сегнетовой соли. Если проводилась предварительная экстракция мешающих веществ, то для анализа берут весь объем раствора, полученного из 50 мл первоначальной пробы. Сегнетову соль тогда не прибавляют. К смеси приливают 10 мл 10%-ного раствора едкого натра и хорошо перемешивают. Экстрагируют 5 мл рабочего раствора дитизона в четыреххлористом углероде. После разделения слоев слой органического растворителя сливают в другую делительную воронку. Экстракцию повторяют с 5 мл раствора дитизона и потом еще порциями по 3 мл этого раствора до тех пор, пока экстракт не станет бесцветным. Соединенные экстракты дважды промывают встряхиванием с 20 мл 2%-ного раствора едкого натра и затем дважды дистиллированной водой. После этого экстракт фильтруют через маленький фильтр в мерную колбу емкостью 25 мл, фильтр промывают малым количеством четыреххлористого углерода и объем раствора дополняют четыреххлористым углеродом до метки. Измеряют оптическую плотность экстракта по отношению к четыреххлористому углероду. Одновременно определяют оптическую плотность холостого определения с 50 мл дважды дистиллированной воды и вычитают ее из оптической плотности пробы.
Растворы дитизоната разлагаются под действием света, поэтому определение рекомендуется проводить в полутемном помещении.
Расчет. Содержание кадмия (x) в мг/л вычисляют по формуле

где c - концентрация кадмия по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 | 1,0 - 2,0 |
Округление, мг/л | 0,01 | 0,02 | 0,05 | 0,1 |
Общие положения
18.1. Естественное содержание калия в поверхностных и подземных водах зависит от геологических условий в бассейне и бывает обыкновенно ниже содержания натрия. Содержание в водах калия может быть увеличено смывами с площадей, обрабатываемых для сельского хозяйства, и спуском некоторых видов сточных вод.
Для определения калия приводятся три метода, из которых наилучшим является фотометрия пламени. На пламенном фотометре можно определять от 0,1 мг/л калия; колориметрическим методом с применением гексанитрокобальтиата можно определить более 1 мг калия в пробе; аргенометрически с тетрафенилборатом можно определить от 0,1 мг калия в объеме пробы, взятой для анализа.
Пробы с низким содержанием калия отбирают в полиэтиленовые бутыли. Можно пользоваться также бутылями из стекла при условии, что анализ производится не позднее чем через 1 сутки после отбора пробы. Пробы не консервируют.
Результаты определений выражают в миллиграммах или в миллиграмм-эквивалентах калия на 1 л воды: 1 мг K+ = 0,0256 мг-экв K+; 1 мг-экв K+ = 39,10 мг K+.
Определение методом фотометрии пламени
18.2. Соединения щелочных и щелочноземельных металлов характерно окрашивают несветящуюся часть пламени. При помощи фильтра, решетки или призмы можно выделить участок спектра специфического или максимального излучения. Характеристической спектральной линией для калия является линия с длиной волны 770 нм.
Мешающие влияния
18.3. При содержании натрия более 25 мг/л и кальция более 50 мг/л результаты определения калия получаются завышенными. В таких случаях калибровочные кривые для калия надо строить по стандартам, содержащим соответствующие количества натрия и кальция.
Аппаратура
18.4. Пламенный фотометр с фильтром для выделения спектральной линии  и принадлежности к нему.
и принадлежности к нему.
и принадлежности к нему.Реактивы
18.5. Хлорид калия, стандартный раствор:
а) запасной: 1,9067 г KCl ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и доводят ею объем до 1 л при 20 °C. Хранят в полиэтиленовой склянке: 1 мл раствора содержит 1,000 мг K+;
б) рабочий: приготовляют, разбавляя в нужной степени запасной раствор.
Ход определения
18.6. Определение проводят с профильтрованной пробой. Измерения производят согласно указаниям руководства для пользования прибором.
Расчет. Содержание ионов калия в мг/л (x) или в мг-экв (y) вычисляют по формулам
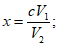

где c - содержание калия, найденное по калибровочной кривой, мг/л;
V1 - объем пробы после разбавления, мл;
V2 - объем пробы, взятой для разбавления, мл.
Округление результатов
Диапазон, мг/л | 0,1 - 0,2 | 2,0 - 5,0 | 5,0 - 10,0 | 10,0 - 20,0 |
Округление: | ||||
мг/л | 0,1 | 0,2 | 0,5 | 1 |
мг-экв | 0,002 | 0,005 | 0,01 | 0,02 |
Колориметрическое определение с гексанитрокобальтиатом
18.7. Колориметрическое определение калия основано на окислении бихроматом малорастворимого гексанитрокобальтиата натрия и калия, выделенного в осадок. Интенсивность окраски раствора бихромата после окисления обратно пропорциональна содержанию калия. Результаты зависят от температуры, от продолжительности окисления и от последовательности проведения операций. В 10 мл объема пробы можно определить 1,0 - 7,0 мг калия, применяя кюветы длиной 5 см; пробу можно концентрировать выпариванием.
Мешающие влияния
18.8. Определению мешают ионы аммония, которые также частично выделяются в виде гексанитрокобальтиата аммония. Кремнекислота может мешать вследствие образования ею мути, появляющейся при выпаривании пробы. Окислению мешают органические вещества. При их наличии отобранную в требуемом объеме пробу минерализуют выпариванием досуха с 2 мл концентрированной серной кислоты и 1 - 2 мл концентрированной азотной кислоты. Полученный сухой остаток растворяют при нагревании в небольшом количестве дистиллированной воды, раствор фильтруют, собирая его количественно в центрифужную пробирку, и дополняют объем дистиллированной водой примерно до 10 мл.
Аппаратура
18.9. Платиновые или кварцевые чашки.
Центрифуга.
Пробирки, емкостью 25 мл.
Фотометр, фильтр  .
.
.Кюветы длиной 5 см или серия цилиндров Несслера.
Реактивы
18.10. Азотная кислота, примерно 1 н. раствор: 64 мл HNO3 ч.д.а., концентрированной, разбавляют дистиллированной водой, доводят объем до 1 л.
Азотная кислота, примерно 0,01 н. раствор: приготовляют, разбавляя однонормальный раствор.
Гексанитрокобальтиат натрия, 20%-ный раствор: 10 г Na3[Co(NO2)6] растворяют в 50 мл дистиллированной водой; перед употреблением раствор фильтруют. Каждый раз применяют свежеприготовленный раствор.
Бихромат калия, 0,1 н. раствор для окисления: 4,9 г K2Cr2O7 ч.д.а. растворяют в дистиллированной воде и доводят объем до 1 л.
Серная кислота ч.д.а., концентрированная.
Хлорид калия, стандартный раствор. Приготовление см. стр. 110; 1 мл раствора содержит 1,000 мг K+.
Калибровочная кривая
18.11. В ряд центрифужных пробирок отмеривают 0 - 1,00 - 2,00 - 3,00 - 4,00 - 5,00 - 6,00 - 7,00 мл стандартного раствора и доводят объемы дистиллированной водой до 10,00 мл. Полученные растворы с концентрациями 0 - 100 - 200 - ... - 700 мг K+ в 1 л обрабатывают описанным ниже способом. Они используются в качестве стандартных растворов цветовой шкалы для визуального колориметрирования или для построения калибровочной кривой. Из величины оптической плотности холостого опыта вычитают ее значение для стандартных растворов и строят график зависимости оптической плотности от содержания калия в мг/л.
Ход определения
18.12. В центрифужную пробирку отмеривают пипеткой 10,0 мл прозрачной пробы, содержащей от 100 до 700 мг калия в 1 л. Пробы с меньшим содержанием калия концентрируют выпариванием в платиновой чашке на водяной бане с таким расчетом, чтобы в полученных 10,0 мл содержалось от 1,0 до 7,0 мг калия. Помутневшие при концентрировании вывариванием пробы центрифугируют или фильтруют.
Перемешивая смесь, на холоду добавляют в нее 1 мл 1 н. раствора азотной кислоты и 5 мл раствора гексанитрокобальтиата, после чего оставляют ее стоять точно 2 ч. Выпавший осадок выделяют центрифугированием в течение 10 мин и осторожно сливают прозрачную жидкость. В пробирку с осадком приливают 15 мл примерно 0,01 н. раствора азотной кислоты и хорошо перемешивают смесь. Повторяют центрифугирование и сливают прозрачный раствор. Добавляют, перемешивая смесь, 10 мл раствора бихромата калия и 5 мл концентрированной серной кислоты. Смесь охлаждают до комнатной температуры, количественно переносят в мерную колбу емкостью 100 мл и доводят объем дистиллированной водой до метки. Параллельно проводят холостое определение с дистиллированной водой. Измеряют оптическую плотность, вводят поправку на холостой опыт с дистиллированной водой и по калибровочной кривой находят содержание калия или же сравнивают интенсивность окраски с серией приготовленных таким же методом стандартов в цилиндрах Несслера.
ИС МЕГАНОРМ: примечание. Текст дан в соответствии с официальным текстом документа. |
Расчет. Содержание ионов калия в мг/л (x) или в мг-экв/л вычисляют по формулам:


где c - содержание (концентрация) калия, найденное по калибровочной кривой или путем сравнения, мг/л;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 10 - 20 | 20 - 50 | 50 - 100 | 100 - 200 |
Округление: | ||||
мг | 1 <*> | 2 <*> | 5 <*> | 10 <*> |
мг/экв | 0,02 | 0,05 | 0,1 | 0,2 |
--------------------------------
Аргентометрическое определение в виде тетрафенилбората
18.13. Калий выделяют из раствора в виде нерастворенного тетрафенилбората калия. Осадок растворяют в ацетоне и калий определяют аргентометрически.
Мешающие влияния
18.14. Определению мешают ионы  , Rb+, Cs+, Ag+, Tl+, Hg2+. Мешающее влияние аммонийных ионов устраняют или кипячением пробы, подщелоченной NaOH, или прокаливанием сухого остатка, полученного выпариванием (см. стр. 111).
, Rb+, Cs+, Ag+, Tl+, Hg2+. Мешающее влияние аммонийных ионов устраняют или кипячением пробы, подщелоченной NaOH, или прокаливанием сухого остатка, полученного выпариванием (см. стр. 111).
Содержание ионов Na+, Ca2+, Mg2+, Mn2+, Zn2+, Cl-,  не мешает определению даже в количестве, в 2000 раз превышающем содержание ионов K+, если осаждение проводят с не очень концентрированным раствором (содержащим не более 50 мг K+ в 1 мл).
не мешает определению даже в количестве, в 2000 раз превышающем содержание ионов K+, если осаждение проводят с не очень концентрированным раствором (содержащим не более 50 мг K+ в 1 мл).
Аппаратура
18.15. Фарфоровый или стеклянный тигель для фильтрования с плотной фильтрующей пластинкой.
Колба для отсасывания с предохранительной склянкой.
Водоструйный насос.
Реактивы
18.16. Соляная кислота, ч.д.а., разбавленная (1:1).
Тетрафенилборат натрия, 0,1 М раствор для осаждения: 34,2 г тетрафенилбората растворяют в дистиллированной воде, приливают 20 мл 10%-ного раствора Al2(SO4)3, дополняют до 1 л и перемешивают. После нескольких часов отстаивания раствор фильтруют через слой окиси алюминия для хроматографии. Реактив в течение нескольких недель не теряет своих свойств.
Промывной раствор: к 1 л дистиллированной воды приливают 30 мл 0,1 М раствора тетрафенилбората натрия и 10 мл ледяной уксусной кислоты.
Смешанный индикатор (эозин + n-диметиламиноазобензол + светлая зелень "licht  "): 300 мг эозина и 5 мг n-диметиламиноазобензола (метиловый желтый) растворяют в 150 мл ацетона. К этому раствору при помешивании приливают раствор 350 мг светлой зелени (натриевая соль диэтилдибензилдиаминотрифенилкарбинолтрисульфоновой кислоты) в 50 мл дистиллированной воды.
"): 300 мг эозина и 5 мг n-диметиламиноазобензола (метиловый желтый) растворяют в 150 мл ацетона. К этому раствору при помешивании приливают раствор 350 мг светлой зелени (натриевая соль диэтилдибензилдиаминотрифенилкарбинолтрисульфоновой кислоты) в 50 мл дистиллированной воды.
Бромид калия, 0,01 н. раствор: 1,19 г KBr ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и разбавляют до 1 л.
Нитрат серебра, 0,01 н. раствор: см. стр. 233.
Ход определения
18.17. Отмеряют такой объем пробы, чтобы в нем содержалось 0,1 - 20 мг калия, и разбавляют до 50 мл дистиллированной водой. Затем пробу подкисляют до  , прибавляя несколько капель разбавленной соляной кислоты. Из раствора при комнатной температуре выделяют осадок прибавлением значительного избытка 0,1 М раствора тетрафенилбората натрия. Сосуд, в котором проводят осаждение, помещают на 5 - 15 мин в холодную воду (не превышать указанное время, так как реактив в кислой среде разрушается). Осадок отфильтровывают через тигель с плотной фильтрующей пластинкой, перенося его в тигель с помощью промывного раствора, и промывают в тигле 2 - 3 раза несколькими миллилитрами промывного раствора, тщательно обмывая им стенки тигля. Таким же способом промывают осадок и стенки тигля еще 1 - 2 раза, возможно меньшим количеством дистиллированной воды. Промывание нужно проводить тщательно, чтобы устранить и следы реактива, и галогениды, иначе результаты получатся повышенными. Затем осадок в тигле растворяют, пропуская через тигель при отсасывании, приблизительно 50 мл ацетона; раствор собирают в колбу для титрования емкостью 200 мл. Ацетон приливают малыми дозами, после растворения осадка тщательно ополаскивают ацетоном стенки и дно тигля, а также резиновую прокладку в горле колбы. К ацетоновому раствору приливают 10,0 мл 0,01 н. раствора бромида калия и несколько капель смешанного индикатора. Титруют 0,01 н. раствором нитрата серебра до перехода окраски индикатора из зеленой в красно-фиолетовую. Одновременно определяют расход титрованного раствора на 10,0 мл раствора KBr с тем же индикатором.
, прибавляя несколько капель разбавленной соляной кислоты. Из раствора при комнатной температуре выделяют осадок прибавлением значительного избытка 0,1 М раствора тетрафенилбората натрия. Сосуд, в котором проводят осаждение, помещают на 5 - 15 мин в холодную воду (не превышать указанное время, так как реактив в кислой среде разрушается). Осадок отфильтровывают через тигель с плотной фильтрующей пластинкой, перенося его в тигель с помощью промывного раствора, и промывают в тигле 2 - 3 раза несколькими миллилитрами промывного раствора, тщательно обмывая им стенки тигля. Таким же способом промывают осадок и стенки тигля еще 1 - 2 раза, возможно меньшим количеством дистиллированной воды. Промывание нужно проводить тщательно, чтобы устранить и следы реактива, и галогениды, иначе результаты получатся повышенными. Затем осадок в тигле растворяют, пропуская через тигель при отсасывании, приблизительно 50 мл ацетона; раствор собирают в колбу для титрования емкостью 200 мл. Ацетон приливают малыми дозами, после растворения осадка тщательно ополаскивают ацетоном стенки и дно тигля, а также резиновую прокладку в горле колбы. К ацетоновому раствору приливают 10,0 мл 0,01 н. раствора бромида калия и несколько капель смешанного индикатора. Титруют 0,01 н. раствором нитрата серебра до перехода окраски индикатора из зеленой в красно-фиолетовую. Одновременно определяют расход титрованного раствора на 10,0 мл раствора KBr с тем же индикатором.
, прибавляя несколько капель разбавленной соляной кислоты. Из раствора при комнатной температуре выделяют осадок прибавлением значительного избытка 0,1 М раствора тетрафенилбората натрия. Сосуд, в котором проводят осаждение, помещают на 5 - 15 мин в холодную воду (не превышать указанное время, так как реактив в кислой среде разрушается). Осадок отфильтровывают через тигель с плотной фильтрующей пластинкой, перенося его в тигель с помощью промывного раствора, и промывают в тигле 2 - 3 раза несколькими миллилитрами промывного раствора, тщательно обмывая им стенки тигля. Таким же способом промывают осадок и стенки тигля еще 1 - 2 раза, возможно меньшим количеством дистиллированной воды. Промывание нужно проводить тщательно, чтобы устранить и следы реактива, и галогениды, иначе результаты получатся повышенными. Затем осадок в тигле растворяют, пропуская через тигель при отсасывании, приблизительно 50 мл ацетона; раствор собирают в колбу для титрования емкостью 200 мл. Ацетон приливают малыми дозами, после растворения осадка тщательно ополаскивают ацетоном стенки и дно тигля, а также резиновую прокладку в горле колбы. К ацетоновому раствору приливают 10,0 мл 0,01 н. раствора бромида калия и несколько капель смешанного индикатора. Титруют 0,01 н. раствором нитрата серебра до перехода окраски индикатора из зеленой в красно-фиолетовую. Одновременно определяют расход титрованного раствора на 10,0 мл раствора KBr с тем же индикатором.Расчет. Содержание ионов калия в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где a - объем 0,01 н. раствора AgNO3, израсходованного на титрование пробы, мл;
b - объем 0,01 н. раствора AgNO3, израсходованного на титрование 10,0 мл 0,01 н. раствора KBr, мл;
V - объем пробы, взятой для анализа, мл;
k - поправка для приведения нормальности раствора AgNO3 к точно 0,01 М.
Округление результатов. Результаты округляют так же, как при колориметрическом определении калия.
Общие положения
19.1. Соли кальция постоянно входят в состав подземных и поверхностных вод. Их содержание определяется геологическими условиями водоносных слоев. Содержание растворенного кальция в воде и изменение его концентрации зависят в естественных условиях обычно от равновесия углекислых солей и двуокиси углерода. В очень жестких водах при нарушении углекислотного равновесия и уменьшении концентрации двуокиси углерода может произойти выделение карбоната кальция. При отборе проб таких вод надо для определения кальция отбирать отдельную пробу и нейтрализовать ее "щелочность" добавлением соляной кислоты. На природную концентрацию кальция в реках может иметь влияние спуск сточных вод.
Для определения содержания кальция в питьевых, поверхностных и сточных водах применяют комплексонометрическое титрование. Комплексонометрическим титрованием пробы объемом 100 мл можно определить от 1 мг/л и выше кальция.
Пробы не консервируют.
Результат определения выражается в миллиграмм-эквивалентах или миллиграммах кальция в 1 л воды: 1 мг-экв Ca2+ = 20,04 мг Ca2+; 1 мг Ca2+ = 0,0499 мг-экв Ca2+.
Комплексонометрическое определение с комплексоном III
19.2. Комплекс иона кальция с анионом этилендиаминтетрауксусной кислоты устойчив в сильнощелочной среде при  , а комплекс ионов магния в этой среде разрушается и магний выделяется в виде гидроокиси. При титровании раствором комплексона III отсутствие иона кальция обнаруживается мурексидом. Раствор комплекса мурексида (пурпурата аммония) с кальцием окрашен в красный цвет, свободная форма индикатора - в фиолетовый. При прибавлении нафтолового зеленого Б переход окраски мурексида становится более отчетливым. Из грязновато-зеленой окраски он переходит через серую и слабо-красную в чисто-синюю. При определении в объеме 100 мл титруемой воды индикаторная поправка равна только 0,02 мл, 0,05 М титрованного раствора комплексона III.
, а комплекс ионов магния в этой среде разрушается и магний выделяется в виде гидроокиси. При титровании раствором комплексона III отсутствие иона кальция обнаруживается мурексидом. Раствор комплекса мурексида (пурпурата аммония) с кальцием окрашен в красный цвет, свободная форма индикатора - в фиолетовый. При прибавлении нафтолового зеленого Б переход окраски мурексида становится более отчетливым. Из грязновато-зеленой окраски он переходит через серую и слабо-красную в чисто-синюю. При определении в объеме 100 мл титруемой воды индикаторная поправка равна только 0,02 мл, 0,05 М титрованного раствора комплексона III.
, а комплекс ионов магния в этой среде разрушается и магний выделяется в виде гидроокиси. При титровании раствором комплексона III отсутствие иона кальция обнаруживается мурексидом. Раствор комплекса мурексида (пурпурата аммония) с кальцием окрашен в красный цвет, свободная форма индикатора - в фиолетовый. При прибавлении нафтолового зеленого Б переход окраски мурексида становится более отчетливым. Из грязновато-зеленой окраски он переходит через серую и слабо-красную в чисто-синюю. При определении в объеме 100 мл титруемой воды индикаторная поправка равна только 0,02 мл, 0,05 М титрованного раствора комплексона III.Мешающие влияния
19.3. Если в отобранной пробе выпал в осадок карбонат кальция и встряхиванием его нельзя снова перевести в раствор, то надо склянку опорожнить, растворить остаток соли на стенках приблизительно в 2 мл разбавленной (1:5) соляной кислоты и пробу налить обратно в склянку. Происходящим при этом увеличением объема пробы, если она превышает 200 мл, можно пренебречь. В подготовленной таким образом пробе нельзя производить другие определения, например: pH, щелочности, хлоридов, окисляемости, БПК, марганца, растворенных и взвешенных веществ.
При анализе вод, содержащих значительные количества растворенных органических веществ, коллоидов и нерастворимых частиц, пробу минерализуют. Взятый объем пробы выпаривают досуха и остаток прокаливают. Выпаривание и прокаливание повторяют еще 2 или 3 раза после прибавления нескольких капель концентрированной соляной кислоты и концентрированной азотной кислоты. В заключение прибавляют 5 мл соляной кислоты (1:1), смесь нагревают и разбавляют дистиллированной водой до объема около 50 мл. Раствор фильтруется горячим, чашку и фильтр ополаскивают разбавленной (1:50) соляной кислотой. Фильтрат нейтрализуют 1 н. раствором едкого натра и пробу приводят к нужному объему.
При высоком содержании в пробе фосфатов (75 мг/л и больше) после доведения pH до 12 - 13 выпадает фосфат кальция. Влияние фосфатов, присутствующих в концентрации выше 75 мг/л, можно устранить предварительным разбавлением пробы.
Определению мешают медь в концентрациях, превышающих 2 мг/л, двухвалентное и трехвалентное железо, если его больше 20 мг/л, марганец в концентрациях 10 мг/л, цинк, свинец, алюминий и олово (больше 5 мг/л). Их влияние устраняется прибавлением сульфида или цианида (см. стр. 55). Взвешенные вещества удаляют фильтрованием или центрифугированием.
Реактивы
19.4. Едкий натр, приблизительно 1 н. раствор: 40 г едкого натра ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л.
Мурексид, твердая смесь: а) 0,2 г мурексида (пурпурата аммония) смешивают со 100 г хлорида натрия ч.д.а. или 100 г сульфата натрия ч.д.а. и растирают в тонкий порошок;
б) 0,2 г мурексида и 0,5 г нафтолового зеленого Б смешивают со 100 г хлорида натрия ч.д.а. и растирают в тонкий порошок.
Комплексон III, 0,05 М раствор (приготовление см. стр. 56).
Ход определения
19.5. В колбу для титрования отбирают пипеткой 100 мл пробы, содержащей не больше 15 мг кальция или меньшее количество пробы, доведенной до объема 100 мл дистиллированной водой. При анализе кислых проб их нейтрализуют добавлением едкого натра в количестве, эквивалентном кислотности пробы. При анализе проб, щелочность которых превышает 6 мг-экв/л, прибавляют эквивалентное количество 0,1 н. соляной кислоты, кипятят 1 мин и охлаждают. Потом прибавляют 2 мл приблизительно 1 н. раствора едкого натра и от 0,1 до 0,2 г смеси индикатора с солью или смеси индикатора с солью и нафтоловым зеленым Б, после чего медленно титруют раствором комплексона III до появления интенсивной фиолетовой окраски (когда применяют смесь мурексида с нафтоловым зеленым Б до чисто-синей окраски).
Расчет. Содержание кальций-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где a - расход 0,05 М титрованного раствора комплексона III, мл;
k - поправка для приведения концентрации раствора комплексона III к точно 0,05 М;
V - количество пробы, взятой для определения, мл;
100 - объем, до которого доведена проба;
20,04 - эквивалент Ca2+-иона.
Округление результатов
Диапазон, мг/л | 1,0 - 20,0 | 20,0 - 50,0 | 50,0 - 100,0 | 100,0 - 200,0 |
Округление: | ||||
мг/л | 0,5 | 1,0 | 2,0 | 5,0 |
мг-экв/л | 0,02 | 0,05 | 0,10 | 0,20 |
Общие положения
20.1. Магниевые соли являются постоянной составной частью подземных и поверхностных вод. Содержание их определяется геологическими условиями в водоносных слоях. Концентрация магниевых солей обыкновенно не превышает концентрацию кальциевых солей.
Содержание магния в питьевых и поверхностных водах может быть вычислено по результатам определения жесткости воды и содержания в ней кальция. В отсутствие кальциевых солей магний определяют комплексонометрическим титрованием.
Пробы не консервируются.
Результаты определения выражаются в миллиграмм-эквивалентах или миллиграммах магния на 1 л воды: 1 мг-экв Mg2+ = 12,16 мг Mg2+; 1 мг Mg2+ = 0,08446 мг-экв Mg2+.
Расчет содержания магния
20.2. Концентрацию магния вычисляют по разности между израсходованными объемами титрованного раствора комплексона III на определение жесткости и на определение кальция (вариант А) или непосредственно по найденным значениям жесткости и концентрации кальция (вариант Б).
Расчет. Вариант А. Содержание ионов магния в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где a - расход 0,05 М раствора комплексона III при определении жесткости, мл;
b - расход 0,05 М раствора комплексона III при определении кальция, мл;
V1 - объем пробы, взятой на определение жесткости, мл;
V2 - объем пробы, взятой на определение кальция, мл;
k - поправка для приведения концентрации раствора комплексона III к точно 0,05 М;
0,05 - молярность раствора комплексона III;
12,16 - эквивалент Mg2+-иона при ацидиметрическом титровании.
Вариант Б. Содержание магний-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
x = 12,16(a - b); y = (a - b),
где 12,16 - эквивалент Mg2+-иона;
a - общая жесткость, мг-экв/л;
b - кальциевая жесткость, мг-экв/л.
Округление результатов
Диапазон, мг/л | 0,5 - 10,0 | 10,0 - 20,0 | 20,0 - 50,0 | 50,0 - 100,0 |
Округление, мг/л | ||||
мг | 0,20 | 0,50 | 1,0 | 2,0 |
мг-экв | 0,02 | 0,05 | 0,1 | 0,2 |
Комплексонометрическое определение с комплексоном III
20.3. После отделения кальция в виде его оксалата содержание магния определяют титрованием раствором комплексона III в среде аммиака и хлорида аммония с индикатором эриохромчерным T (вариант А). Можно применить двойное титрование раствором комплексона III (вариант Б). Сначала пробу титруют с мурексидом, затем изменяют pH и титруют с эриохромчерным T. По варианту Б первым титрованием находят содержание кальция, вторым - содержание магния. Весь расход комплексона III на титрование показывает жесткость воды. При обработке 100 мл пробы можно определить магний в концентрациях от 1 до 60 мг/л.
Мешающие влияния
20.4. Определению мешают катионы, реагирующие с индикатором или с комплексоном III. Устранение этого влияния описано при определении жесткости. Поскольку для анализа применяется фильтрат после выделения кальция в виде оксалата, в растворе отсутствуют железо, алюминий, марганец и стронций. Мешающие влияния кадмия, меди, свинца и цинка устраняются добавлением раствора цианида или сульфида, как это описано при определении жесткости.
Мешающее влияние растворенных, коллоидных и нерастворимых органических веществ устраняется минерализацией пробы, выполняемой, как это описано при определении кальция.
Реактивы
20.5. Оксалат аммония, раствор для осаждения: 35 г хлорида аммония ч.д.а. и 1,5 г оксалата аммония (NH4) C2O4·H2O отдельно растворяют в дистиллированной воде, оба раствора смешивают, прибавляют 3,5 мл концентрированного раствора аммиака и доводят дистиллированной водой до 250 мл.
Аммиак + хлорид аммония, буферный раствор, pH = 10 (см. стр. 56).
Комплексон III, 0,05 М раствор, приготовление см. стр. 56.
Эриохромчерный T, смесь индикатора с солью, приготовление см. стр. 56.
Едкий натр, 1 н. раствор, приготовление см. стр. 35.
Мурексид, смесь с солью, приготовление см. стр. 116.
Соляная кислота, 1 н. раствор: 85 мл концентрированной HCl ч.д.а. разбавляют дистиллированной водой до 1 л.
Ход определения
20.6. Вариант А. К 100 мл пробы или к меньшему ее объему, доведенному до 100 мл дистиллированной водой, прибавляют 25 мл раствора оксалата аммония и смесь хорошо перемешивают. Выпавший в осадок оксалат кальция отфильтровывают через сухой плотный фильтр (синяя лента) в сухой сосуд. Из фильтрата отбирают пипеткой 50 мл в колбу для титрования, прибавляют 2,5 мл буферного раствора и около 0,1 г твердой смеси индикатора. Титруют раствором комплексона III сначала до перехода красной окраски в фиолетовую, потом медленно, знергично перемешивая, до появления синего окрашивания. Израсходованный объем раствора комплексона III умножают на 2,5 и получают расход реактива на 100 мл пробы.
Если анализируют соединенные фильтраты после определения кальция, то сначала доводят их объем до 100 мл или же переносят фильтраты в мерную колбу, разбавляют до метки и отбирают аликвотную часть соответственно количеству присутствующего магния. После прибавления 5 мл буферного раствора и около 0,1 г индикатора титруют раствором комплексона III до перехода фиолетовой окраски в синюю.
Вариант Б. В колбу для титрования отмеряют 100 мл пробы. Щелочность нейтрализуют прибавлением эквивалентного количества 0,1 н. соляной кислоты. Потом прибавляют 2 мл раствора едкого натра, 0,1 - 0,2 г смеси мурексида с солью и сразу титруют раствором комплексона III до появления интенсивного фиолетового окрашивания. Израсходованный объем отвечает содержанию кальция. После окончания титрования раствор нейтрализуется 1 н. раствором соляной кислоты (1,5 - 2,5 мл). Разложение мурексида ускоряют нагреванием (или прибавлением капли бромной воды). Затем прибавляют 5 мл буферного раствора, около 0,1 г смеси эриохромчерного с солью и титруют до перехода фиолетового окрашивания в синее.
Расчет. Содержание магний-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
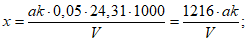

где a - расход 0,05 М раствора комплексона III при титровании с эриохромчерным, мл;
V - объем первоначальной пробы, взятой для определения, мл;
k - поправка для приведения концентрации раствора комплексона III к точно 0,05 М;
24,31 - эквивалент Mg2+-иона в комплексонометрическом титровании;
12,16 - эквивалент Mg2+-иона в ацидиметрическом титровании.
Округление результатов. Результаты округляют, как при определении магния расчетом.
Весовое определение
20.7. Метод заключается в осаждении магния фосфатом аммония и прокаливании при 1100 °C. Образовавшийся фосфат магния и аммония переводится в пирофосфат магния. Метод пригоден для определения 0,3 - 60 мг магния.
Определение проводят в фильтрате после определения кальция в виде оксалата.
Мешающие влияния
20.8. Определению мешают мутность, коллоидные органические вещества, силикаты, железо, марганец и алюминий.
Аппаратура
20.9. Электрическая печь, 1100 °C.
Реактивы
20.10. Фосфат аммония двухзамещенный (NH4)2HPO4, 10%-ный раствор.
Аммиак ч.д.а., концентрированный раствор.
Аммиак ч.д.а., разбавленный (1:99) раствор.
Соляная кислота ч.д.а., разбавленная (1:9).
Соляная кислота ч.д.а., разбавленная (1:99).
Метиловый красный, 0,1%-ный раствор.
Ход определения
20.11. Фильтрат после определения кальция, доведенный до объема примерно 150 мл, подкисляют после прибавления нескольких капель метилового красного соляной кислотой до красной окраски. При постоянном перемешивании прибавляют примерно 10 мл раствора фосфата аммония, нейтрализуют концентрированным раствором аммиака до желтой окраски индикатора. При постоянном перемешивании, во время которого палочка не должна прикасаться к стенкам стакана, прибавляют еще 5 мл концентрированного раствора аммиака. Затем раствору дают отстояться не менее 4 ч и фильтруют количественно через фильтр "синяя лента". Осадок на фильтре растворяют примерно в 50 мл горячей разбавленной (1:9) соляной кислоты и раствор собирают в стакан, в котором производилось осаждение. Фильтр тщательно промывают горячей разбавленной (1:99) соляной кислотой. Объем фильтрата доводят до 150 мл, прибавляют 1 - 2 мл раствора фосфата аммония и охлаждают до комнатной температуры. После прибавления нескольких капель раствора метилового красного вновь осаждают MgNH4PO4 добавлением концентрированного раствора аммиака. Смесь оставляют не менее чем на 4 ч и фильтруют через плотный фильтр (синяя лента). Осадок на фильтре несколько раз промывают разбавленным раствором аммиака. После высушивания во взвешенном тигле фильтр с осадком сжигают, прокаливают при 1100 °C до постоянного веса и взвешивают образовавшийся пирофосфат магния.
Расчет. Содержание магния в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
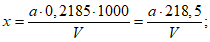

где a - вес Mg2P2O7, мг;
0,2185 - коэффициент пересчета с Mg2P2O7 на Mg, мг/л;
17,97 - коэффициент пересчета с Mg2P2O7 на Mg, мг-экв/л;
V - объем пробы, мл.
Округление результатов
Диапазон, мг/л | 2,0 - 50,0 | 50,0 - 100,0 | 100,0 - 200,0 | 200,0 - 500,0 |
Округление: | ||||
мг | 0,5 | 1,0 | 2,0 | 5,0 |
мг-экв | 0,05 | 0,10 | 0,20 | 0,50 |
Общие положения
21.1. Марганец присутствует в подземных водах обычно в растворенной форме в виде двухвалентных ионов (из-за отсутствия кислорода), а в поверхностных водах - в нерастворенной форме в виде гидроокисей высших степеней валентности. В подземных и поверхностных водах концентрация марганца зависит от геологических условий.
Большое количество марганца бывает в сточных водах от обработки руд, в стоках металлургических заводов, в шахтных водах и в некоторых сточных водах химической промышленности.
Для определения общего содержания всех форм марганца в питьевых и поверхностных водах приводится колориметрический метод, основанный на окислении персульфатом солей марганца в азотнокислой среде в присутствии катализатора нитрата серебра до перманганат-ионов, интенсивность окраски которых сравнивают с интенсивностью окраски стандартного раствора перманганата калия. Интенсивность окраски пропорциональна концентрации марганца.
Определение марганца рекомендуется производить сразу после отбора пробы ввиду его быстрого окисления и выпадения в осадок. В тех случаях, когда марганец не может быть определен сразу после отбора пробы, последнюю консервируют добавлением 5 мл концентрированной азотной кислоты на 1 л пробы.
Без изменения объема пробы можно определять марганец при содержании его от 0,05 до 10 мг в 1 л. Для определения меньших концентраций марганца в питьевых и поверхностных водах необходимо увеличить его концентрацию в пробе упариванием. Чувствительность метода - 0,01 мг/л.
Колориметрическое определение
Мешающие влияния
21.2. Определению мешают хлориды и органические вещества. Высокое содержание железа (III), меди, никеля и бихроматов мешает окраской своих ионов.
Метод пригоден при содержании в воде хлоридов до 100 мг в анализируемом объеме воды. Мешающее влияние хлоридов устраняется добавлением к пробе сульфата ртути. Помехи от присутствия в воде органических веществ устраняют нагреванием пробы и добавлением указанного ниже количества персульфата аммония.
Фильтрование исследуемой воды не рекомендуется во избежание задержания малорастворимых соединений марганца на фильтре.
При визуальном сравнении растворов количественно определить влияние мутности и цветности анализируемой воды невозможно. С целью снижения влияния мутности и цветности на определение марганца исследуемую воду можно разбавить дистиллированной водой.
При определении марганца на электрофотоколориметре мешающее влияние мутности и цветных катионов исключают двойным измерением: после определения оптической плотности окрашенной пробы окраску от перманганат-иона уничтожают добавлением в кювету капли 30%-ного раствора перекиси водорода и повторяют измерение оптической плотности; содержание марганца вычисляют по разности.
Аппаратура
21.3. Цилиндры Геннера.
Электрофотоколориметр, светофильтр  .
.
.Набор кювет.
Реактивы
21.4. Комбинированный реактив: растворяют 75 г сульфата ртути HgSO4 в 400 мл концентрированной азотной кислоты и 200 мл дистиллированной воды. Добавляют 200 мл 85%-ной фосфорной кислоты H3PO4 и 0,035 г нитрата серебра AgNO3, раствор охлаждают и доводят дистиллированной водой до 1 л.
Персульфат аммония (NH4)2S2O8, 30%-ный раствор.
Перманганат калия, стандартные растворы 0,01 н. и 0,05 н.
Перекись водорода, 30%-ный раствор.
Калибровочная кривая
21.5. В ряд мерных колб емкостью 100 мл наливают 70 - 80 мл дистиллированной воды, 5 мл комбинированного реактива, 3,5 мл раствора персульфата аммония и различные количества стандартного 0,01 н. раствора KMnO4 (1 мл 0,011 н. раствора KMnO4 содержит 0,11 мг Mn): 0,1; 0,3 - 0,5 - 0,8 - 1,0 - 1,5 мл и т.д., что после доведения объемов жидкости в колбах дистиллированной водой до 100 мл соответствует концентрациям марганца 0,11 - 0,33 - 0,55 - 0,88 - 1,1 - 1,65 и т.д. мг/л.
Ход определения
21.6. К 70 - 80 мл анализируемой воды добавляют 5 мл комбинированного реактива, 3,5 мл раствора персульфата аммония, доводят до кипения и кипятят 1 - 2 мин на электрической плитке до установления постоянной окраски. (Длительное кипячение пробы приводит к разложению избытка персульфата и потере окраски, такой же эффект дает медленный подогрев пробы.)
Снимают колбу с плитки и через 1 мин охлаждают под водопроводным краном. Доводят пробу до 100 мл дистиллированной водой и перемешивают.
Интенсивность окраски определяют визуально, сравнением в цилиндрах Геннера (вариант А) или на электрофотоколориметре (вариант Б).
Вариант А. Приблизительно 50 мл приготовленного раствора переливают в цилиндр Геннера N 1. Для приготовления раствора сравнения в мерную колбу емкостью 100 мл наливают 70 - 80 мл дистиллированной воды, 5 мл комбинированного реактива, 3,5 мл раствора персульфата аммония, перемешивают и добавляют из микробюретки по каплям 0,01 н. или 0,05 н. раствор KMnO4 до получения приблизительно такой же окраски, как в цилиндре Геннера N 1.
Доводят объем жидкости в мерной колбе дистиллированной водой точно до 100 мл и приблизительно 50 мл переливают в цилиндр Геннера N 2.
Просматривают столбы жидкостей в обоих цилиндрах сверху вниз и выпускают через кран одного из цилиндров часть более окрашенной жидкости до выравнивания окраски.
Расчет. Содержание марганца (x) в мг/л рассчитывают по формуле

где a - количество 0,01 н. или 0,05 н. раствора KMnO4, пошедшее на приготовление раствора сравнения в цилиндре Геннера N 2, мл;
b - содержание марганца в стандартных растворах KMnO4; для 0,01 н. KMnO4 b = 0,11 мг/мл; для 0,05 н. KMnO4 b = 0,055 мг/мл;
k - поправочный коэффициент для приведения нормальности растворов KMnO4 к точно 0,01 н. и 0,05 н.;
V - объем анализируемой воды, взятый для определения, мл;
h1 - высота столба жидкости в цилиндре Геннера N 1, см;
h2 - высота столба жидкости в цилиндре Геннера N 2, см.
Вариант Б. Определяют величину оптической плотности в кюветах с толщиной слоя 2 - 5 см (в зависимости от интенсивности окраски) и по калибровочной кривой находят содержание марганца.
Расчет. Содержание марганца (II) в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где c - концентрация Mn, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для анализа, мл;
27,47 - эквивалент Mn2+-иона;
100 - объем, до которого разбавлена проба, мл.
Округление результатов
Диапазон, мг/л | 0,02 - 1,00 | 1,00 - 2,00 | 2,0 - 5,0 | 5,0 - 10,0 |
Округление: | ||||
мг/л | 0,02 | 0,05 | 0,1 | 0,2 |
мг-экв/л | 0,0002 | 0,001 | 0,002 | 0,05 |
Общие положения
22.1. Присутствие меди в подземных водах связано с составом горных пород. В поверхностных водах медь в большинстве случаев присутствует в результате загрязнения их промышленными сточными водами. Источником меди является также коррозия медных или содержащих медь металлических частей, соприкасающихся с водами, например трубопроводов для питьевой и производственной воды, сооружений для охлаждения воды при оборотных системах водоснабжения. В питьевых и в поверхностных водах можно обнаружить также медь, внесенную альгицидными (уничтожающими водоросли) реагентами.
Медь может находиться в воде в виде двухвалентных катионов или же быть связанной в форме комплексов - медноцианистых, меднотартратных и др. В нерастворимой форме медь встречается в виде сульфида, гидроокиси, карбоната.
Колориметрический метод с экстракцией диэтилдитиокарбамината меди хлороформом и прямые определения с тетраэтилтиурамдисульфидом и с оксальдигидразидом рекомендуется для анализа питьевых и поверхностных вод, а после минерализации пробы - и для анализа сточных вод, содержащих медь в концентрациях от 0,01 до 5 мг в 1 л.
Пробы, содержащие цианиды, не следует консервировать. Отобранные пробы консервируют, добавляя 5 мл концентрированной азотной кислоты на 1 л пробы, что препятствует адсорбции меди на стенках сосуда, в котором проба хранится. Особенно необходимо консервировать пробы, в которых содержание меди меньше 1 мг/л. Если требуется раздельное определение растворенной и нерастворенной меди, то перед консервированием пробу фильтруют и определяют медь в нефильтрованной и в фильтрованной пробах.
Результаты определений выражаются в миллиграммах меди на 1 л воды.
Качественное определение
Реакция с диэтилдитиокарбаминатом
22.2. К 20 мл пробы прибавляют 1 мл раствора цитрата аммония, 2 мл раствора комплексона III и 2 мл раствора диэтилдитиокарбамината натрия (приготовление реактивов см. ниже, стр. 126). Появление коричневого осадка является доказательством присутствия не менее десятых долей миллиграмма меди в 1 л; коричневая муть появляется уже при содержании 0,05 мг/л меди. После добавления 2 мл хлороформа и встряхивания слой хлороформа должен приобрести желто-коричневую окраску. Эту окраску можно заметить уже при содержании 0,02 мг меди в 1 л. Помутнение раствора с образованием белого осадка или желтой окраски не является доказательством присутствия меди. Медь, связанная в комплексы с цианидами, не может быть открыта описанным способом. Для ее обнаружения пробу в таких случаях необходимо предварительно подготовить, как это изложено ниже.
Реакция с тетраэтилтиурамдисульфидом
22.3. Прибавляют к 20 мл пробы несколько капель концентрированной соляной кислоты, 20 мл этилового спирта и 1 мл 0,3%-ного раствора тетраэтилтиурамдисульфида в этиловом спирте. После перемешивания появляется окрашивание (от желтого до коричневого цвета). Этим способом можно еще определить присутствие 0,002 мг Cu в 1 л пробы.
Реакция с оксальдигидразидом
22.4. Прибавляют к 20 мл пробы 3 мл раствора цитрата аммония, 1 мл концентрированного раствора аммиака, 1 мл раствора оксальдигидразида и 3 мл раствора ацетальдегида. После прибавления каждого реактива раствор перемешивают. В присутствии меди через 10 мин возникает красно-фиолетовая окраска. Чувствительность - 0,001 мг меди в 1 л воды.
Колориметрическое определение
с диэтилдитиокарбаминатом натрия
22.5. Ионы меди реагируют с диэтилдитиокарбаминатом с образованием коричневого, нерастворимого в воде диэтилдитиокарбамината меди, который легко растворяется в хлороформе, окрашивая последний в желто-коричневый цвет. Интенсивность окраски прямо пропорциональна концентрации меди. Медь экстрагируют из аммиачной среды, содержащей цитрат аммония и комплексон III, который маскирует большинство металлов, вступающих в реакцию с указанным реактивом.
Описанным методом, анализируя 100 мл пробы, можно определить от 0,05 до 1,0 мг меди в 1 л.
Мешающие влияния
22.6. Комплексные цианиды должны быть до определения разрушены выпариванием пробы после добавления к ней 0,5 мл разбавленной (1:1) серной кислоты и 5 мл концентрированной азотной кислоты. Выпаривание проводят в вытяжном шкафу. К остатку после выпаривания добавляют 1 мл концентрированной соляной кислоты и снова выпаривают досуха. Полученный остаток растворяют в дистиллированной воде, подогревая смесь, если понадобится. Затем фильтруют через стеклянную фильтрующую пластинку и анализируют фильтрат описанным ниже методом.
Такая подготовка пробы может служить и для исключения мешающего определению влияния небольших количеств органических веществ. Пробы с высоким содержанием органических веществ, мешающих реакции, необходимо минерализовать выпариванием с азотной и серной кислотами.
В аммиачной среде, содержащей комплексон III, в реакции с диэтилдитиокарбаминатом вступают помимо меди висмут, серебро и ртуть. Комплексы серебра и ртути практически бесцветны и на определение меди не влияют. Комплекс висмута имеет желтый цвет и частично оказывает влияние на результаты анализа. Оптическая плотность хлороформового экстракта диэтилдитиокарбамината висмута соответствует примерно одной пятой оптической плотности диэтилдитиокарбамината меди такой же концентрации.
При высоких концентрациях кальция и магния и других элементов, вступающих в реакцию с диэтилдитиокарбаминатом, следует соответственно увеличить количество добавляемого комплексона III. Не менее половины содержащегося в смеси комплексона III должно остаться в форме свободного.
Аппаратура
22.7. Фотометр, светофильтр  .
.
.Кюветы длиной 1 - 5 см.
Реактивы
22.8. Диэтилдитиокарбаминат натрия, 1%-ный раствор: 1 г диэтилдитиокарбамината натрия ч.д.а. растворяют в небольшом количестве дистиллированной воды, фильтруют и доводят объем раствора дистиллированной водой до 100 мл; раствор хранят в склянке из темного стекла; он устойчив около месяца.
Аммиак ч.д.а., концентрированный раствор разбавляют в отношении 1:1.
Хлороформ ч.д.а. или очищенный дистилляцией.
Цитрат аммония ч.д.а., 40%-ный раствор.
Комплексон III, примерно 0,1 М раствор: 37,2 г комплексона III (двухнатриевой соли этилендиаминтетрауксусной кислоты) растворяют в дистиллированной воде и доводят ею объем до 1 л.
Сульфат меди, стандартный раствор:
а) запасной: 0,200 г медной фольги или медной проволочки ч.д.а. растворяют в 10 мл разбавленной (1:1) азотной кислоты. После растворения приливают 1 мл концентрированной серной кислоты ч.д.а. и выпаривают до появления паров сорной кислоты. Объем раствора дополняют при 20 °C до 1 л; 1 мл раствора содержит 0,200 мг меди;
б) рабочий I: 250 мл запасного раствора разбавляют до объема 1 л. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,050 мг Cu;
в) рабочий II: 20,0 мл стандартного рабочего раствора I разбавляют до 1 л. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,001 мг меди.
Калибровочная кривая
22.9. В делительные воронки отмеривают 0 - 2,0 - 5,0 - 10,0 - 20,0 - 40,0 - 60,0 - 100,0 мл стандартного рабочего раствора II и доводят объемы дважды дистиллированной водой до 100 мл, получая серию стандартов с содержанием 0 - 0,02 - 0,05 - 0,010 - ... - 1,00 мг меди в 1 л. Стандарты эти обрабатывают описанным ниже способом. Из величин измеренных оптических плотностей вычитают оптическую плотность холостого определения и полученные разности наносят на график против соответствующих концентраций меди в мг/л.
Для приготовления всех реактивов, для разбавления пробы и для мытья использованной посуды применяют воду, дважды перегнанную в приборе из стекла (бидистиллят).
Ход определения
22.10. В делительную воронку емкостью 250 - 500 мл помещают 100 - 250 мл пробы либо первоначальной, либо предварительно разбавленной или сконцентрированной выпариванием так, чтобы в этом объеме содержалось 0,005 - 0,1 мг меди. Очень кислые или сильнощелочные пробы нейтрализуют соответственно разбавленным раствором едкого натра или соляной кислоты. На каждые 100 мл пробы добавляют 5 мл раствора цитрата аммония, 10 мл раствора комллексона III, 10 мл раствора аммиака и 10 мл хлороформа. Эту смесь перемешивают около 1 мин и встряхивают. Слою хлороформа дают собраться на дне воронки, после чего его выпускают и удаляют. Если он окрашен, то экстракцию продолжают новыми порциями хлороформа по 10 мл до тех пор, пока полученный экстракт не будет бесцветным. Затем к водному раствору добавляют 10 мл хлороформа (из цилиндра, в который заранее было отмерено 24 мл хлороформа) и 10 мл раствора диэтилдитиокарбамината натрия. Примерно двухминутным встряхиванием проводят экстракцию, дают спокойно отделиться хлороформному слою и выпускают его, фильтруя затем через малый бумажный фильтр (белая лента), в мерную колбу емкостью 25 мл. Вливают в делительную воронку 2 мл хлороформа и тотчас же выпускают их в колбу, споласкивая, таким образом, как отверстие крана, так и трубку воронки. После этого экстрагируют новой порцией в 5 мл хлороформа, встряхивая в течение 2 мин, и выпускают хлороформ после отделения слоев в ту же мерную колбу через тот же фильтр. Как и при предшествовавшем экстрагировании, отверстие крана и трубки воронки прополаскивают примерно 1 мл хлороформа.
Экстрагирование и прополаскивание повторяют с 5 и с 1 мл хлороформа. Собранные вместе в мерной колбе экстракты доводят хлороформом до метки и перемешивают.
Измерение оптической плотности проводят быстро, так как хлороформ испаряется. Из измеренной величины вычитают оптическую плотность холостого определения с дистиллированной водой.
Расчет. Содержание меди (x) в мг/л вычисляют по формуле

где c - содержание меди (концентрация), найденное по калибровочной кривой, мг;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,00 | 1,0 - 2,0 |
Округление, мг/л | 0,01 | 0,02 | 0,05 | 0,1 |
Более высокие и более низкие концентрации округляются аналогично, согласно приведенной схеме.
Общие положения
23.1. Нормальное содержание натрия в поверхностных и подземных водах соответствует геологическим условиям в бассейне и в водоносных слоях. Содержание это увеличивается спуском в водоемы хозяйственно-фекальных и производственных сточных вод.
Наиболее рационально определять натрий методом фотометрии пламени. Приведен также метод определения натрия осаждением его цинкуранилацетатом с весовым и объемным окончаниями.
Пробы воды с малыми концентрациями натрия отбирают в полиэтиленовые бутыли. Можно пользоваться для этого также бутылями из стекла, из которого вода не извлекает щелочных металлов, при условии, что анализ производится не позднее чем через одни сутки после отбора пробы. Пробы не консервируют.
Результаты определений выражают в миллиграммах или в миллиграмм-эквивалентах натрия в 1 л воды; 1 мг Na+ = 0,0435 мг-экв Na+; 1 мг-экв Na+ = 22,99 мг Na+.
Определение методом фотометрии пламени
23.2. Соединения щелочных и щелочноземельных металлов характерно окрашивают несветящее пламя. При помощи фильтра, решетки или призмы можно выделить участок спектра из специфического или же максимального излучения. Для натрия характерна спектральная линия с длиной волны 589 нм.
Метод пригоден для прямого определения от 1 до 100 мг натрия в 1 л воды.
Мешающие влияния
23.3. Если содержание натрия больше, чем содержание калия, и если отношение содержания натрия к содержанию кальция превышает 1:10, то калий и кальций определению практически не мешают. Для устранения мешающего влияния катионов, спектральные линии которых близки к спектральной линии натрия, целесообразно построение калибровочных кривых со стандартными растворами, содержащими мешающие компоненты в известных концентрациях.
Аппаратура
23.4. Пламенный фотометр с фильтром для выделения линии с длиной волны 589 нм, с решеткой или призмой, снабженный необходимыми принадлежностями.
Реактивы
23.5. Хлорид натрия, стандартный раствор:
а) запасной: 2,5421 г NaCl ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и доводят ее объем до 1 л. Раствор хранят в полиэтиленовой бутыли; 1 мл раствора содержит 1,000 мг Na+;
б) рабочий: приготовляют разбавлением запасного раствора.
Ход определения
23.6. Для определения используют профильтрованную пробу. Измерения производят способом, изложенным в приложенном к прибору наставлении.
Расчет. Содержание натрий-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
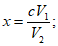

где c - содержание натрия, найденное по калибровочной кривой, мг/л;
V1 - объем пробы после разбавления, мл;
V2 - объем пробы, взятой для разбавления, мл;
22,99 - эквивалент Na+-иона.
Округление результатов
Диапазон, мг/л | 0,1 - 2,0 | 2,0 - 5,0 | 5,0 - 10,0 | 10,0 - 20,0 |
Округление: | ||||
мг | 0,1 | 0,2 | 0,5 | 1 |
мг-экв | 0,005 | 0,01 | 0,02 | 0,05 |
Определение осаждением в виде натрий-цинкуранилацетата
23.7. Для определения натрия используется малая растворимость ацетата натрия, цинка и уранила NaZn(UO2)3(C2H3O2)9·6H2O, выделяющегося из пробы после прибавления десятикратного избытка смеси ацетата цинка с ацетатом уранила. Выпавший осадок тройного ацетата или взвешивают (метод А), или растворяют и определяют в полученном растворе цинк комплексонометрически (вариант Б). Взвешиванием определяют 1 - 8 мг, титрованием 0,1 - 2 мг натрия в объеме пробы, взятой для анализа.
Мешающие влияния
23.8. Определению мешает литий, осаждающийся подобно натрию. Калий при содержании его в анализируемой пробе до 25 мг на результаты определения не влияет. Весовому определению мешают кремнекислота и фосфаты. Кремнекислота увлекается в осадок; фосфаты выпадают в осадок в форме фосфорноураниловой соли. Исключение этих влияний предусматривается в ходе определения.
Мешают определению также некоторые органические кислоты, влияние которых устраняют выпариванием нужного объема пробы досуха на водяной бане, в платиновой чашке, с 1 - 2 мл концентрированной азотной кислоты. Остаток в чашке растворяют в теплом состоянии в дистиллированной воде. Таким же образом удаляют из пробы муть, если мутность нельзя устранить ни фильтрованием, ни центрифугированием.
Аппаратура
23.9. Посуда из стойкого стекла, из которого не выщелачиваются щелочные металлы.
Фарфоровый или стеклянный тигель с фильтрующим дном средней пористости.
Вакуум-насос.
Реактивы
23.10. Цинкуранилацетат, осаждающий раствор: 10 г ацетатуранила (UO2)(C2H3O2)2·2H2O ч.д.а. растворяют в 54 мл горячей дистиллированной воды, содержащей 1,7 мл ледяной уксусной кислоты; 30 г ацетата цинка Zn(C2H3O2)2·2H2O ч.д.а. растворяют в 52 мл горячей дистиллированной воды, содержащей 1 мл ледяной уксусной кислоты. После растворения солей (при нагревании) оба раствора смешивают и прибавляют к их смеси небольшое количество хлорида натрия. Смесь основательно перемешивают и оставляют стоять 24 ч, после чего ее фильтруют. Раствор хранят в склянке из стойкого, невыщелачивающегося стекла.
Этиловый спирт, 96%-ный, насыщенный на холоду осадком цинк-натрийуранилацетата. Осадок специально для этой цели приготовляют, добавляя в раствор цинкуранилацетата хлорид натрия.
Этиловый эфир ч.д.а.
Для комплексонометрического определения дополнительно требуется: эриохром черный T, смесь с NaCl. Приготовление см. стр. 56.
Карбонат аммония ч.д.а., твердый.
Раствор аммиака, приблизительно 3 н.: 200 мл 25%-ного раствора аммиака ч.д.а. разбавляют дистиллированной водой до 1 л.
Комплексон III (трилон Б), 0,01 М титрованный раствор: 3,721 г комплексона III (двузамещенной натриевой соли этилендиаминтетрауксусной кислоты) ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л. Поправку к нормальности раствора определяют по стандартному раствору сульфата цинка, полученному отвешиванием 0,6537 г металлического цинка и растворением в серной кислоте, как описано на стр. 152; 1 мл этого раствора отвечает 1 мл точно 0,01 М раствора комплексона III.
Ход определения
23.11. Весовое определение. В стакане на 20 мл, в небольшой платиновой или кварцевой чашке упаривают соответствующий объем пробы примерно до 1 мл. Сгущенная таким путем проба может содержать не больше чем 8 мг натрия и 25 мг калия. После охлаждения прибавляют 10 мл осадительного раствора, перемешивают смесь и оставляют на 1 ч. Выпавший осадок фильтруют, пропуская его под небольшим разрежением через предварительно взвешенный фильтрующий тигель. Осадок в тигле промывают 5 раз осадительным раствором порциями по 2 мл и тоже 5 раз спиртом - порциями по 2 мл. После отсоса жидкости осадок смачивают несколькими каплями эфира и продолжают отсасывание еще 10 - 15 мин. Тигель оставляют 10 - 15 мин постоять в футляре весов, после чего его взвешивают.
При присутствии кремниевой кислоты или фосфатов взвешенный тигель-фильтр переносят опять в отсасывающий прибор. Осадок растворяют в 100 мл дистиллированной воды, прибавляемой малыми дозами. Тигель 5 раз промывают порциями спирта по 2 мл, смачивают эфиром, высушивают отсасыванием жидкости и взвешивают так же, как в предшествующем случае. Разность между двумя взвешиваниями дает массу натрий-цинкуранилацетата.
Расчет. Содержание натрий-ионов в мг/л (x) или в мг-экв (y) вычисляют по формулам:


где m - масса осадка, мг;
0,01495 - коэффициент пересчета с NaZn(UO2)3(C2H3O2)9·6H2O;
22,99 - эквивалентный вес Na+-иона;
V - объем пробы, взятой для анализа, мл.
Объемное определение
23.12. В стакане емкостью 20 мл, небольшой платиновой или кварцевой чашке упаривают соответствующий объем пробы, содержащей 0,1 - 2 мг натрия, примерно до 1 мл, затем охлаждают. Прибавляют 10 мл осаждающего реактива, смесь перемешивают и оставляют ее на 1 ч. Выпавший осадок отфильтровывают, пропуская раствор под небольшим разрежением через фильтрующий тигель.
Остатки осадка переносят из стакана (чашки) в фильтрующий тигель, споласкивая их пятью порциями по 2 мл осаждающего реактива. Содержимое тигля затем промывают 5 раз спиртом, порциями по 2 мл. Промытый осадок растворяют в 5 - 10 мл дистиллированной воды, прибавляемой по частям. Раствор собирают в подставленную колбу емкостью 50 мл. Затем прибавляют примерно 0,1 - 0,2 г твердого карбоната аммония, 0,5 мл раствора аммиака и столько индикатора, чтобы раствор явственно окрасился в красный цвет, после чего титруют 0,01 М раствором комплексона III до перехода окраски из красной в синюю.
Расчет. Содержание натрий-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где a - объем израсходованного 0,01 М раствора комплексона III, мл;
k - поправочный коэффициент для приведения концентрации раствора комплексона III к точно 0,01 М;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 5,0 - 10,0 | 10,0 - 20,0 | 20,0 - 50,0 | 50,0 - 100,0 |
Округление: | ||||
мг | 0,5 | 1 | 2 | 5 |
мг-экв | 0,02 | 0,05 | 0,1 | 0,2 |
Общие положения
24.1. Никель не является, как правило, составной частью природных вод. Его присутствие обусловлено загрязнением промстоками.
Никель находится в воде в растворимой форме в виде двухвалентного катиона или комплексных ионов, наиболее часто - в виде цианидного комплекса и в нерастворимой форме - в виде цианида, сульфида, карбоната или гидроокиси никеля.
Для определения никеля приводится колориметрический метод, применимый при концентрациях никеля 0,2 - 20 мг/л. Для определения никеля в более высоких концентрациях (выше 5 мг/л) приведен весовой метод с диметилглиоксимом.
Пробы консервируют добавлением 2 - 5 мл концентрированной азотной кислоты на 1 л пробы. Если требуется отдельно определить никель в растворенной и нерастворенной формах, пробу до консервирования фильтруют и определяют общее содержание никеля и содержание его в растворе. Пробы, содержащие цианиды, нельзя консервировать.
Результаты приводятся в миллиграммах никеля на 1 л воды.
Качественное определение
24.2. К 20 мл пробы прибавляют примерно 2 мл 3%-ного раствора перекиси водорода и раствор аммиака до явно щелочной реакции. Смесь кипятят и выпавший осадок отфильтровывают. К фильтрату добавляют 2 мл 1,2%-ного раствора диметилглиоксима и раствор нагревают до кипения. Если присутствует малое количество никеля, появляется желтая окраска, если концентрация никеля превышает 2,5 мг/л, выпадает красный осадок.
Колориметрическое определение с диметилглиоксимом
24.3. Ионы никеля в слабоаммиачной среде в присутствии сильного окислителя вступают в реакцию с диметилглиоксимом с образованием комплексного соединения красного цвета. Интенсивность окраски пропорциональна концентрации никеля. Прямым способом можно определить 0,2 - 5 мг/л никеля.
Мешающие влияния
24.4. Железо, хром и медь перед определением необходимо из раствора удалить. Для удаления железа из раствора к 100 мл пробы добавляют 2 мл 3%-ной перекиси водорода, нагревают и осаждают гидроокись железа прибавлением раствора аммиака. Выпавший осадок отфильтровывают.
Хроматы и бихроматы восстанавливают несколькими каплями этилового спирта после подкисления пробы серной кислотой. Трехвалентный хром затем отделяют, осаждая его разбавленным (1:4) раствором аммиака. Если присутствуют только ионы трехвалентного хрома, сразу осаждают раствором аммиака и отфильтровывают выпавший осадок.
Медь осаждают пропусканием сероводорода в пробу, подкисленную соляной кислотой до pH = 2. Осадок отфильтровывают и сероводород удаляют из раствора кипячением.
Аппаратура
24.5. Фотометр, светофильтр  .
.
.Кюветы толщиной 1 - 5 см или цилиндры Несслера емкостью 100 мл.
Реактивы
24.6. Бром, насыщенный водный раствор.
Раствор аммиака ч.д.а., концентрированный.
Диметилглиоксим, 1,2%-ный раствор: 1,2 г натриевой соли диметилглиоксима ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Сульфат никеля, стандартный раствор:
а) запасной: 1,000 г никелевой проволоки ч.д.а. растворяют в 15 мл примерно 35%-ной азотной кислоты ч.д.а., добавляют 5 мл разбавленной (1:3) серной кислоты ч.д.а. и выпаривают до появления белых паров серной кислоты. Остаток растворяют и раствор доливают дистиллированной водой до 1 л при 20 °C; 1 мл раствора содержит 1 мг никеля;
б) рабочий I: 25,0 мл запасного стандартного раствора разбавляют дистиллированной водой до 1 л. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,025 мг никеля;
в) рабочий II: 100,0 мл рабочего раствора I разбавляют до 500 мл дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,005 мг никеля.
Калибровочная кривая
24.7. В мерные колбы емкостью 100 мл отмеривают 0 - 2,5 - 5,0 - 10,0 - 20,0 - 30,0 - 40,0 - 50,0 мл рабочего стандартного раствора II и разбавляют каждый раствор дистиллированной водой до 50 мл. Таким способом получают растворы с концентрацией 0 - 0,25 - 5,0 мг/л никеля. Эти растворы обрабатывают описанным ниже способом. Измеряют оптическую плотность, вводят поправки на холостое определение и полученные значения наносят на график против соответствующих концентраций никеля в мг/л.
При работе в цилиндрах Несслера приготовляют стандартные растворы указанных выше концентраций, отмеряя 0 - 2,5 - 5,0 - 10,0 - 15,0 - 20,0 - 25,0 до 50,0 мл рабочего раствора II в цилиндры Несслера емкостью 100 мл, и обрабатывают их одновременно с пробами описанным ниже способом.
Ход определения
24.8. В мерную колбу или цилиндр Несслера емкостью 100 мл помещают 50 мл пробы, если надо предварительно разбавленной или сконцентрированной так, чтобы в этом объеме содержалось 0,01 - 0,025 мг никеля. Прибавляют 10 мл насыщенного раствора брома и смесь перемешивают. После того прибавляют 12 мл раствора аммиака, 4 мл раствора диметилглиоксима и доливают дистиллированной водой до метки. Одновременно проводят холостой опыт с 30 мл дистиллированной воды. Из величины оптической плотности пробы вычитают оптическую плотность холостого определения.
Аналогичным способом проводят определение в цилиндрах Несслера, добавляя требуемые реактивы одновременно к пробе и к стандартным растворам.
Расчет. Содержание никеля (x) в мг/л вычисляют по формуле

где c - концентрация никеля по калибровочной кривой или полученная сравнением со стандартом, мг/л;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,20 - 0,50 | 0,50 - 1,0 | 1,0 - 2,0 | 2,0 - 5,0 |
Округление, мг/л | 0,02 | 0,05 | 0,1 | 0,2 |
Более высокие и более низкие концентрации округляются аналогичным образом.
Весовое определение с диметилглиоксимом
24.9. Ионы никеля в аммиачной среде с диметилглиоксимом образуют светло-красное соединение, не растворимое в воде, которое после отделения фильтрованием и высушивания при 110 - 120 °C имеет постоянный состав.
Мешающие влияния
24.10. Мешающее влияние оказывает двухвалентное железо. К пробе, отмеренной для анализа, прибавляют около 2 мл 3%-ной перекиси водорода и кипятят до полного разложения перекиси. Затем прибавляют винную кислоту, маскирующую железо и другие элементы, образующие в аммиачной среде осадки.
Если проба содержит цианиды, к отмеренному объему пробы прибавляют 5 мл концентрированной азотной кислоты и 0,5 мл разбавленной (1:1) серной кислоты и выпаривают до появления белого дыма. Работают под тягой! Остаток растворяют в воде, раствор разбавляют и обрабатывают описанным выше способом.
Аппаратура
24.11. Фильтрующий тигель.
Колба для отсасывания с предохранительной склянкой.
Вакуумный насос.
Сушильный шкаф (110 - 120 °C).
Реактивы
24.12. Диметилглиоксим, 1,2%-ный раствор, приготовление см. стр. 133.
Аммиак ч.д.а., разбавленный раствор (1:5).
Винная кислота ч.д.а.
Соляная кислота ч.д.а., разбавленная (1:5).
Ход определения
24.13. К 100 мл пробы прибавляют раствор аммиака до появления слабого запаха и смесь нагревают до кипения. После охлаждения до 50 - 60 °C приливают раствор диметилглиоксима до прекращения образования осадка. Раствор при этой температуре оставляют на водяной бане примерно полчаса. Затем прибавлением еще 3 мл раствора диметилглиоксима убеждаются в полноте осаждения никеля. Осадок отфильтровывают через фильтрующий тигель и промывают горячей дистиллированной водой. После этого осадок растворяют в малом количестве разбавленной соляной кислоты и промывают тигель горячей водой. Фильтрат собирают в стакан и объем его доводят до 100 - 150 мл дистиллированной водой. Прибавляют раствор аммиака до слабого запаха и при температуре 50 - 60 °C прибавляют еще раствор диметилглиоксима до полного выпадения осадка. При этой температуре оставляют смесь примерно на полчаса. После этого фильтруют через взвешенный фильтрующий тигель и осадок на фильтре промывают горячей дистиллированной водой. Тигель с осадком сушат примерно 3 ч при температуре 110 - 120 °C. После охлаждения в эксикаторе взвешивают диметилглиоксимат никеля.
Расчет. Содержание никеля (x) в мг/л вычисляют по формуле

где m - масса диметилглиоксимата никеля, мг;
V - объем пробы, взятой для определения, мл;
0,2032 - коэффициент пересчета с диметилглиоксимата никеля на никель.
Округление результатов
Диапазон, мг/л | 5,0 - 10,0 | 10,0 - 20,0 | 20,0 - 50,0 |
Округление, мг/л | 0,2 | 0,5 | 1 |
Общие положения
25.1. Ртуть встречается изредка в шахтных водах и в сточных водах промышленности. Ртуть присутствует в воде чаще всего в растворимой форме в виде недиссоциированных молекул, ионов Hg2+ и иногда  . Ртуть присутствует в воде также в нерастворимой форме и в составе комплексных соединений.
. Ртуть присутствует в воде также в нерастворимой форме и в составе комплексных соединений.
Для определения ртути рекомендуется экстракционный колориметрический метод с применением дитизона. Этот метод почти специфичен для ртути и его можно применять для определения как очень малых ее концентраций порядка сотых долей миллиграмма, так и десятков миллиграммов ртути в 1 л.
Пробы не консервируют, только при длительном хранении к ним прибавляют 1 мл азотной кислоты на 1 л анализируемой воды для предотвращения адсорбции ионов или соединений ртути на стенках сосуда.
Результаты определений приводятся в миллиграммах ртути на 1 л.
Качественное определение
25.2. В пробирку наливают примерно 10 мл пробы, прибавляют 5 мл ацетатного буферного раствора, 5 мл раствора комплексона III, 5 мл раствора роданида калия и после перемешивания - примерно 2 мл хлороформного раствора дитизона (приготовление всех реактивов см. стр. 138). Содержимое пробирки хорошо встряхивают. Если присутствует ртуть, окраска хлороформного раствора перейдет из зеленой в оранжевую.
Колориметрическое определение с дитизоном
25.3. При экстрагировании водного раствора соли ртути раствором дитизона в хлороформе появляется оранжевый дитизонат ртути, растворимый в хлороформе. В слабокислой среде ацетатного буфера и в присутствии комплексона III и роданида калия реакция ртути с дитизоном практически специфична. Избыток свободного дитизона из экстракта удаляют встряхиванием с разбавленным раствором аммиака и измеряют оптическую плотность раствора дитизоната ртути в хлороформе.
При обработке 100 мл пробы можно определить ртуть в концентрациях 0,05 - 1 мг/л с точностью +/- 5%. Более низкие концентрации ртути определяются экстрагированием из большого объема пробы (до 500 мл) или после упаривания предварительно подщелоченного раствора.
Мешающие влияния
25.4. В слабокислой среде в присутствии комплексона III и роданида с дитизоном, помимо ртути, вступают в реакцию также трехвалентное золото и, вероятно, двухвалентная платина. Однако эти элементы на практике в водах не встречаются.
Определению могут помешать красители, экстрагируемые хлороформом. Если такие вещества присутствуют, то после прибавления ацетатного буфера, комплексона и роданида экстрагируют малыми порциями хлороформа до тех пор, пока последний экстракт не останется бесцветным. Только тогда экстрагируют раствором дитизона.
Большие количества органических соединений необходимо устранить минерализацией. Минерализацию применяют также в случаях, когда присутствуют органические соединения ртути. Пробу минерализуют кипячением с перманганатом в кислой среде. Подходящее количество пробы (с содержанием 0,005 - 0,1 мг ртути) отмеряют в колбу, снабженную притертой пробкой, и прибавляют 1 мл концентрированной серной кислоты и несколько капель насыщенного раствора перманганата калия. Затем опускают в колбу несколько стеклянных шариков или капилляров и присоединяют ее к охлаждаемому водой обратному холодильнику. Смесь умеренно кипятят. Если раствор обесцветится, то через обратный холодильник прибавляют по каплям еще немного раствора перманганата до тех пор, пока фиолетовая окраска в колбе не останется в течение 15 мин. Большой избыток перманганата вредит определению. Раствор охлаждают, споласкивают обратный холодильник дистиллированной водой в колбу, отключают колбу и по каплям прибавляют насыщенный раствор гидроксиламина сернокислого или гидрозина сернокислого до полного обесцвечивания анализируемого раствора.
Ртуть определяется потом описанным ниже способом.
Определению мешает присутствие сильных окислителей. Их присутствие в пробе обнаруживают прибавлением нескольких капель соляной кислоты, 10%-ного раствора йодида калия и раствора крахмала. Если реакция дает положительный результат, то к приготовленной для определения пробе прибавляют нужное количество насыщенного раствора сульфата гидразина или гидроксиламина.
Сильнокислые и сильнощелочные пробы (к ним относятся и пробы после минерализации или упаривания) следует до определения нейтрализовать разбавленной серной кислотой или разбавленной щелочью (примерно 1 н.) до слабокислой реакции 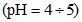 .
.
.Аппаратура
25.5. Фотометр, светофильтр  .
.
.Кюветы толщиной 1 - 2 см.
Реактивы
25.6. Буферный раствор (уксусная кислота + ацетат натрия): 57 мл ледяной уксусной кислоты ч.д.а. и 82 г CH3COONa·3H2O ч.д.а. растворяют в дистиллированной воде и дополняют до 1 л.
Комплексон III, примерно 0,1 н. раствор: 37,2 г двунатриевой соли этилендиаминтетрауксусной кислоты ч.д.а. растворяют в дистиллированной воде и дополняют до 1 л.
Роданид калия, примерно 0,1 н. раствор: 9,72 г роданида калия ч.д.а. растворяют в дистиллированной воде и дополняют до 1 л.
Аммиак, 5%-ный раствор: 200 мл концентрированного раствора аммиака ч.д.а. (примерно 25%-ного) разбавляют до 1 л дистиллированной водой.
Хлороформ ч.д.а. или очищенный перегонкой.
Дитизон, раствор для экстрагирования:
а) запасной: 50 мг дитизона растворяют в 100 мл хлороформа в делительной воронке и прибавляют 200 мл дистиллированной воды и 5 - 10 мл концентрированного раствора аммиака ч.д.а. Смесь хорошо встряхивают примерно 2 мин. После разделения слоев хлороформный слой спускают. В делительную воронку прибавляют 20 мл хлороформа, смесь немного встряхивают, дают постоять и хлороформ снова опускают. Конец делительной воронки высушивают фильтровальной бумагой и в воронку прибавляют 200 мл хлороформа и разбавленную соляную кислоту до отчетливо кислой реакции. Смесь встряхивают, пока выделенный дитизон не перейдет в хлороформ. Хлороформный слой сливают в другую делительную воронку и трижды промывают встряхиванием с 50 мл дистиллированной воды. После этого раствор дитизона сливают в коричневую склянку и сверху осторожно покрывают слоем 0,5 процентов серной кислоты, содержащей 0,5% сульфата гидразина. Раствор хранят в темноте и на холоду. Он устойчив несколько месяцев;
б) рабочий: к 1 объему запасного раствора прибавляют 4 объема хлороформа. Раствор хранят в коричневой бутылке на холоду. Раствор устойчив 14 дней.
Нитрат ртути (II), стандартный раствор:
а) запасной: запасным раствором служит 0,05 н. раствор нитрата ртути (II), применяемый при меркуриметрическом определении хлоридов. Приготовление и определение поправки см. стр. 235; 1 мл раствора содержит 5,0152 k мг ртути;
б) рабочий I: 49,86 k мл 0,05 н. запасного раствора (k - поправка) из бюретки отмеряют в мерную колбу емкостью 1000 мл и доливают до метки дистиллированной водой; 1 мл полученного раствора содержит 0,25 мг ртути;
в) рабочий II: 20,0 мл рабочего стандартного раствора I разбавляют до 1 л дистиллированной водой. Применяют каждый раз свежеприготовленный раствор; 1 мл раствора содержит 0,005 мг ртути.
Калибровочная кривая
25.7. В ряд мерных колб емкостью 250 мл отмеряют 0 - 2,5 - 5,0 - 10,0 - 15,0 - ... - 50,0 мл рабочего стандартного раствора II и колбы доливают дистиллированной водой до меток. Из приготовленных таким способом стандартов с концентрацией 0 - 0,05 - 0,10 - ... - 1,0 мг/л ртути отбирают по 100 мл в делительные воронки и обрабатывают описанным выше способом.
Измеряют оптическую плотность, вводят поправку на холостое определение и полученные значения наносят на график против соответствующих концентраций ртути в мг/л.
Ход определения
25.8. Выбирают делительные воронки, первую - емкостью 250 - 750 мл (по объему обрабатываемой пробы), остальные - по 100 - 150 мл. В первую и вторую воронки помещают по 10 мл буферного раствора, 10 мл раствора комплексона III и 10 мл раствора роданида калия. Во вторую колбу прибавляют еще 20 мл дистиллированной воды. Растворы перемешивают. В третью воронку наливают 50 мл 5%-ного раствора аммиака. Затем в первую воронку отмеряют такой объем пробы, чтобы в нем содержалось 0,005 - 0,1 мг ртути. Прибавляют примерно 10 мл хлороформа, смесь встряхивают и после разделения хлороформный слой отбрасывают. После этого в первую воронку прибавляют 25 мл рабочего раствора дитизона и экстрагируют примерно в течение 2 мин не очень сильным встряхиванием. После разделения слоев смесь еще раз немного встряхивают и дают хорошо отстояться. Экстракт спускают в другую воронку, куда при этом не должно перейти ни малейшего количества водного слоя (отверстие в кране первой воронки остается заполненным хлороформным раствором). Содержимое второй воронки встряхивают примерно 1 мин и дают разделиться слоям. Экстракт спускают в третью воронку и опять встряхивают примерно 1 мин (нужно опять тщательно отделить хлороформный слой от водного). После хорошего разделения хлороформный раствор фильтруют через маленький бумажный фильтр. Первую порцию фильтрата отбрасывают, а остальное собирают в кювету и измеряют оптическую плотность. Аналогично проводят холостой опыт с таким же объемом дистиллированной воды. Полученные значения оптической плотности вычитают из значения, полученного при проведении определения, и по калибровочной кривой определяют содержание ртути.
Расчет. Содержание ртути (x) в мг/л вычисляют по формуле

где c - содержание ртути, найденное по калибровочной кривой, мг;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,010 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,00 |
Округление, мг/л | 0,005 | 0,01 | 0,02 | 0,05 |
Общие положения
26.1. Свинец встречается иногда в подземных водах, он может присутствовать также в питьевой воде, куда он попадает при соприкосновении воды со свинцовым трубопроводом. Источником свинца в поверхностных водах могут служить промышленные сточные воды. Свинец в воде может присутствовать в растворимой форме в виде простых или комплексных ионов. В нерастворимой форме он встречается в виде сульфида, карбоната и сульфата.
Для определения свинца в питьевых и поверхностных водах приводится экстракционный колориметрический метод с дитизоном; этим методом можно определять свинец в концентрациях от сотых долей миллиграмма до целых миллиграммов в 1 л воды.
Пробы консервируют добавлением 3 мл концентрированной азотной кислоты или 2 мл ледяной уксусной кислоты на 1 л пробы. Если необходимо раздельно определить свинец в растворе и в твердой фазе, пробу фильтруют непосредственно после ее отбора и определяют свинец в профильтрованном и в первоначальном растворах. Осадок переводят в раствор выпариванием подходящего объема пробы с азотной кислотой досуха. Остаток затем смачивают 1 мл азотной кислоты и растворяют в дистиллированной воде.
Результаты определения выражаются в миллиграммах свинца в 1 л воды.
Качественное определение
26.2. К 10 мл пробы прибавляют 1 мл 26%-ного раствора тартрата калия и натрия (сегнетовой соли), 0,5 мл 0,5%-ного раствора едкого натра и 0,5 мл 10%-ного раствора цианида калия. (Осторожно, яд!). Смесь перемешивают. Прибавляют 1 мл свежеприготовленного 5%-ного раствора сульфида натрия. В присутствии свинца появляется желтая окраска, а при больших концентрациях - коричневая окраска или коричневый осадок. Чувствительность определения 0,3 мг свинца в 1 л воды.
Колориметрическое определение с дитизоном
26.3. Сущность метода заключается в образовании дитизоната свинца, окрашенного в красный цвет и растворимого в четыреххлористом углероде. Дитизонат свинца экстрагируют при pH - 8 - 9 в цианидной среде, в которой маскируется присутствие большинства металлов, реагирующих с дитизоном. В 100 мл пробы можно определить свинец в концентрации 0,1 - 1,0 мг/л.
Мешающие влияния
26.4. В щелочной среде, содержащей цианид, дитизоном экстрагируются вместе со свинцом таллий, висмут и двухвалентное олово. Олово и висмут удаляют экстрагированием в кислой среде. К пробе после восстановления гидразином (см. ход определения) и после охлаждения прибавляют 20 мл раствора тартрата натрия. pH доводят до 2,5 - 3 винной кислотой, прибавляемой по каплям (проверяют потенциометрически). Пробу затем количественно переносят в делительную воронку и экстрагируют порциями по 5 мл 0,1%-ного хлороформного раствора дитизона до тех пор, пока зеленая окраска дитизона не перестанет изменяться. После этого экстрагируют порциями по 5 мл хлороформа, пока экстракт не останется бесцветным. Хлороформ из пробы удаляют экстрагированием 5 мл четыреххлористого углерода. К водному раствору после экстракции прибавляют 5 капель раствора тимолового синего (см. ниже "Реактивы") и концентрированным раствором аммиака нейтрализуют до появления синего окрашивания индикатора. Этим способом из пробы удаляют вместе с висмутом и оловом также медь, серебро и ртуть.
Определению не мешает присутствие серебра, ртути, меди, мышьяка, сурьмы, алюминия, хрома, никеля, кобальта и цинка в концентрациях, не превышающих двенадцатикратную концентрацию свинца. Мешающее влияние некоторых из этих элементов, если они присутствуют в пятидесятикратной концентрации, устраняют двойной экстракцией. Раствор дитизоната, полученный описанным способом, встряхивают с двумя порциями по 50 мл 1%-ной азотной кислоты. Водные экстракты, содержащие присутствующий свинец, сливают в другую делительную воронку. Слой четыреххлористого углерода промывают, взбалтывая его два раза с порциями по 20 мл дистиллированной воды, промывную воду прибавляют к водному экстракту.
Определению мешает марганец, который при экстрагировании в щелочной среде каталитически ускоряет окисление дитизона кислородом воздуха. Это мешающее влияние устраняется добавлением гидразина к экстрагируемой пробе. Если марганец присутствует а большой концентрации, хорошо пробу экстрагировать дважды способом, описанным выше.
Если присутствует большое количество органических веществ, пробу минерализуют выпариванием досуха с азотной и соляной кислотами. Остаток после выпаривания смачивают азотной кислотой, растворяют в дистиллированной воде и дальше продолжают описанным ниже способом.
Красители, экстрагируемые четыреххлористым углеродом, удаляются предварительной экстракцией пробы этим растворителем. Экстрагируют порциями по 5 мл растворителя до тех пор, пока очередная порция экстракта не останется бесцветной.
Сильные окислители мешают определению, так как окисляют дитизон. Их восстановление гидразином включено в ход определения.
Аппаратура
26.5. Фотометр, светофильтр  .
.
.Кюветы длиной 2 - 5 см.
Реактивы
26.6. Дважды дистиллированная вода. Получают повторной перегонкой в стеклянном приборе. Применяют ее для приготовления реактивов и для разбавления проб.
Фенолфталеин, 0,5%-ный раствор, приготовление см. стр. 36.
Аммиак ч.д.а., концентрированный раствор.
Уксуснокислый гидразин, раствор для восстановления: 15 мл 64%-ного раствора гидразингидрата смешивают с 50 мл уксусной кислоты (ледяной) и разбавляют до 100 мл дистиллированной водой.
Тартрат натрия, 10%-ный раствор: 10 г Na2C4H4O6·2H2O растворяют в дистиллированной воде и раствор разбавляют до 200 мл. Затем экстрагируют примеси малыми количествами раствора дитизона в четыреххлористом углероде до тех пор, пока последний экстракт не окажется бесцветным. Потом промывают водный раствор малыми порциями четыреххлористого углерода до тех пор, пока последняя порция не останется бесцветной.
Винная кислота ч.д.а., 50%-ный раствор.
Дитизон, раствор для экстрагирования в четыреххлористом углероде:
а) запасной: 50 мг дитизона растворяют в 100 мл четыреххлористого углерода в делительной воронке, добавляют 200 мл дистиллированной воды и 5 - 10 мл концентрированного раствора аммиака ч.д.а. Смесь тщательно перемешивают в течение 2 мин. После разделения слоев четыреххлористый углерод сливают. В делительную воронку прибавляют 20 мл четыреххлористого углерода, смесь немного взбалтывают, дают постоять и снова сливают слой органического растворителя. Конец воронки высушивают фильтровальной бумагой и в воронку добавляют 200 мл четыреххлористого углерода и разбавленную соляную кислоту до явно кислой реакции. Смесь встряхивают до тех пор, пока выделенный дитизон не перейдет в четыреххлористый углерод. Слой растворителя сливают в другую воронку и три раза промывают порциями по 50 мл дистиллированной воды. После этого раствор дитизона переливают в коричневую склянку и осторожно наливают сверху 0,5%-ный раствор серной кислоты, содержащий 0,5% сернокислого гидразина NH2NH2·H2SO4.
Раствор хранят на холоду в темном помещении. Он устойчив в течение нескольких месяцев;
б) рабочий: 50 мл запасного раствора дитизона дополняют четыреххлористый углеродом до 100 мл.
Четыреххлористый углерод ч.д.а., или очищенный перегонкой (см. стр. 107).
Тимоловый синий, 0,4%-ный раствор: 0,4 г натриевой соли тимолового синего растворяют в дистиллированной воде и разбавляют до 100 мл. Прибавляют разбавленный раствор NaOH или HCl до правильной окраски перехода pH.
Промывной раствор (NH4OH + KCN + Na2SO3). К 175 мл концентрированного раствора аммиака ч.д.а. прибавляют 15 мл 10%-ного раствора KCN ч.д.а. и 7,5 мл 10%-ного раствора Na2SO3 ч.д.а. (раствор сульфита можно освободить от следов свинца экстрагированием дитизоном тем же способом, какой указан для очистки тартрата натрия). Смесь доливают до 100 мл дистиллированной водой. (Осторожно, яд!).
Соляная кислота ч.д.а., концентрированная.
Нитрат свинца, стандартный раствор:
а) запасной: 0,200 г свинца ч.д.а. растворяют при нагревании в 4 мл разбавленной (1:1) азотной кислоты и доливают до 1 л дистиллированной водой; 1 мл раствора содержит 0,200 мг Pb;
б) рабочий: 10,0 мл запасного стандартного раствора разбавляют до 1 л дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,0020 мг Pb.
Калибровочная кривая
26.7. В стаканы отмеряют 0 - 2,5 - 5,0 - 10,0 - 15,0 - 20,0 - 30,0 - 40,0 и 50,0 мл стандартного рабочего раствора нитрата свинца и дистиллированной водой доводят каждый раствор до 100 мл. Таким образом получают концентрации 0 - 0,050 - 0,10 - ... - 1,00 мг/л свинца. Стандартные растворы обрабатывают описанным ниже способом, включая операцию восстановления. При экстрагировании всех стандартных растворов применяют такое же количество дитизона, какое было применено при экстрагировании наиболее концентрированного из них. Из значений оптической плотности каждого экстракта вычитают значение ее для холостого определения и полученные значения наносят на график против соответствующей концентрации свинца в мг/л.
Ход определения
26.8. В стакан наливают 100 мл пробы, если надо, предварительно разбавленной или упаренной, чтобы содержание свинца в ней было в пределах 0,010 - 0,100 мг. Затем на водяной бане пробу выпаривают примерно до 30 мл и добавляют несколько капель раствора фенолфталеина и раствор аммиака до появления красной окраски. После этого прибавляют 10 мл раствора уксуснокислого гидразина и смесь нагревают при 90 - 95 °C не менее 10 мин. Раствор затем количественно переводят в делительную воронку, прибавляют 10 мл раствора тартрата натрия, 5 капель тимолового синего и добавляют раствор аммиака до появления синей окраски индикатора. Потом приливают 10 мл раствора цианида и прибавлением по каплям винной кислоты доводят pH до значения, при котором окраска индикатора переходит из синей в зеленую. Экстрагируют встряхиванием с 10 мл раствора дитизона в четыреххлористом углероде. При спускании экстракта в другую делительную воронку в нее не должно попасть ни малейшего количества водного раствора (в кране воронки должно остаться немного хлороформного экстракта). Экстрагирование продолжают порциями по 5 мл дитизона до тех пор, пока окраска хлороформного раствора не перестанет изменяться. После этого экстрагируют еще 5 мл чистого четыреххлористого углерода. Все экстракты, собранные во второй воронке, промывают встряхиванием с 20 мл промывного раствора до исчезновения зеленой окраски непрореагировавшего дитизона. Раствор дитизоната свинца спускают в мерную колбу емкостью 50 мл, а водный слой цианида взбалтывают с двумя порциями по 2 мл четыреххлористого углерода, которые потом также сливают в ту же мерную колбу, после чего раствор в колбе доливают четыреххлористым углеродом до метки и перемешивают. Измеряют оптическую плотность. Одновременно проводят холостое определение с дистиллированной водой и таким же объемом раствора дитизона, какой был использован при анализе пробы. Измеряют оптическую плотность холостого определения и вычитают значения этой величины из полученной при определении свинца в пробе, и по калибровочной кривой находят содержание свинца.
Расчет. Содержание свинца (x) в мг/л вычисляют по формуле

где c - концентрация свинца, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой к определению, мл.
Округление результатов
Диапазон, мг/л | 0,020 - 0,050 | 0,050 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 |
Округление, мг/л | 0,002 | 0,005 | 0,01 | 0,02 |
Остальные концентрации округляют аналогично.
Общие положения
27.1. Серебро встречается в некоторых рудничных водах и в сточных водах фотографической промышленности. В этих водах оно может присутствовать как в растворенном, так и нерастворенном виде, большей частью в форме галоидных солей. Серебро из бактерицидных или альгицидных реагентов может быть обнаружено в питьевых и поверхностных водах.
Для определения серебра приводится метод с применением n-диметиламинобензилиденроданина, которым можно прямо определять серебро при содержании его от десятых долей милиграмма в 1 л воды.
Чувствительность определения можно повысить предварительным упариванием пробы.
Пробы консервируют, добавляя по 5 мл концентрированной азотной кислоты на 1 л воды.
Результаты определений выражаются в миллиграммах серебра на 1 л воды.
Качественное определение
27.2. В 10 мл пробы вливают 3 капли ацетонового раствора n-диметиламинобензилиденроданина (приготовление см. ниже) и 5 мл эфира, после чего смесь перемешивают встряхиванием. При присутствии в пробе серебра граница соприкосновения жидкостей окрашивается в красный цвет.
Колориметрическое определение
с n-диметиламинобензилиденроданином
27.3. Ионы серебра реагируют с n-диметиламинобензилиденроданином с образованием красного осадка, остающегося при малых концентрациях серебра в состоянии коллоидного распределения. Коллоид стабилизируют гуммиарабиком (желатиной и т.п.). По интенсивности окраски в некоторых пределах концентраций проводят колориметрическое определение.
Мешающие влияния
27.4. Определение серебра при помощи n-диметиламинобензилиденроданина специфично, причем ему не мешает ни один из обычных ингредиентов вод.
Мешает муть, в которой часто содержатся нерастворенные соединения присутствующего в воде серебра. Муть, так же как и значительные количества органических веществ, устраняют минерализацией пробы. Для этой цели отмеренное количество пробы выпаривают досуха с 1 мл концентрированной серной кислоты и 2 мл концентрированной азотной кислоты. Остаток после выпаривания растворяют в дистиллированной воде; раствор, если надо, фильтруют и продолжают анализ, как изложено ниже.
Таким же образом разрушают цианидные комплексы и нерастворимые галогениды серебра.
Аппаратура
27.5. Фотометр, светофильтр  или набор цилиндров Несслера емкостью 50 мл.
или набор цилиндров Несслера емкостью 50 мл.
или набор цилиндров Несслера емкостью 50 мл.Реактивы
27.6. Тартрат калия и натрия (сегнетова соль), 20%-ный раствор.
Аммиак, 10%-ный раствор.
Желатина, 1%-ный раствор, приготовляют каждый раз заново.
Ацетон ч.д.а.
n-Диметиламинобензилиденроданин, 0,03%-ный раствор в ацетоне. Раствор сохраняют в темной склянке в холодном месте. Он устойчив в течение примерно двух недель.
Сульфат серебра, стандартный раствор:
а) запасной: 1,445 г Ag2SO4 ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде, добавляют 1 мл концентрированной H2SO4 и доводят объем до 1 л дистиллированной водой. 1 мл раствора содержит 1,00 мг;
б) рабочий: 10,0 мл запасного раствора разбавляют дистиллированной водой до 1 л. Применяют каждый раз свежеприготовленный раствор. 1 мл раствора содержит 0,01 мг Ag.
Калибровочная кривая
27.7. Отмеривают в колбы 0 - 0,5 - 1,0 - 2,0 - 4,0 - 6,0 - 8,0 - 10,0 мл рабочего стандартного раствора и доводят объем каждого раствора дистиллированной водой до 50,0 мл, что соответствует содержанию серебра 0 - 0,1 - 0,2 - ... - 2,0 мг/л. Растворы обрабатывают описанным выше способом: измеряют оптическую плотность, вычитают из найденных величин оптической плотности холостого определения и наносят разности на график против соответствующих концентраций серебра в мг/л.
Ход определения
27.8. К 50 мл прозрачной пробы (первоначальной или предварительно сконцентрированной упариванием) добавляют 2 мл раствора сегнетовой соли, 2 мл раствора аммиака и 1 мл раствора желатины. Смесь перемешивают, приливают 0,5 мл раствора n-диметиламинобензилиденроданина и через 5 мин измеряют оптическую плотность или сравнивают появившуюся окраску со стандартами.
Тем же способом проводят холостой опыт с дистиллированной водой. Измеряют оптическую плотность пробы и вычитают оптическую плотность холостого опыта.
Если проба в самом начале была окрашенной, то из величины оптической плотности пробы вычитают значение ее для холостого опыта, проведенного так же, как и анализ пробы, но в котором вместо 0,5 мл указанного реактива добавляют 0,5 мл ацетона.
Расчет. Содержание серебра (x) в мг/л вычисляют по формуле

где c - концентрация серебра, найденная по калибровочной кривой или сравнением со стандартами, мг/л;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,00 | 1,0 - 2,0 |
Округление, мг/л | 0,01 <*> | 0,02 <*> | 0,05 <*> | 0,1 |
--------------------------------
Общие положения
28.1. Хром попадает в природные воды со сточными водами некоторых машиностроительных, химических производств и кожевенных заводов. В них хром может встречаться в виде трехвалентного катиона или в виде анионов: хромат- или бихромат-ионов. Трехвалентная форма хрома устойчива, и в обычных условиях нельзя предполагать окисления трехвалентного хрома (III) до хрома (VI). В растворенном виде хром (III) присутствует только в кислой среде. В нейтральной и щелочной средах он гидролизуется с выделением гидроокиси хрома (III). Комплексообразующие вещества препятствуют гидролизу. Хром (VI) может встречаться в щелочных растворах в виде хромат-ионов, в кислых растворах - в виде бихромат-ионов. В этой форме хром (VI) устойчив в водах, не содержащих восстановителей, если же восстановители присутствуют, то происходит восстановление его до хрома (III). В твердой фазе присутствует преимущественно гидроокись хрома (III).
Для определения хрома приводится колориметрический метод с дифенилкарбазидом, применимый при содержании хрома от 0,05 до 1 мг/л. При анализе проб, содержащих хром в больших концентрациях, пробу надо предварительно разбавить. Ниже описаны методы определения хрома (VI) и общего содержания хрома; содержание хрома (III) находят по разности.
Пробы, особенно с небольшим содержанием хрома, консервируют на месте их отбора, добавляя к ним 3 - 5 мл концентрированной азотной кислоты на 1 л воды.
Пробы, предназначенные для определения растворенной формы, фильтруют как можно скорее после взятия пробы, еще до ее консервирования подкислением. Если требуется раздельное определение хрома (III) и хрома (IV), то пробы анализируют в день их взятия; анализировать пробу позднее (не позже чем на третьи сутки) можно только в тех случаях, когда нет оснований опасаться восстановления хрома (VI) содержащимися в пробе веществами.
Результаты определения всех форм выражаются в миллиграммах хрома в 1 л воды.
Качественное определение
28.2. Определение с едким натром и перекисью водорода (общее содержание хрома). К 10 мл пробы прибавляют 1 мл 30%-ного раствора едкого натра и 1 мл 3%-ной перекиси водорода; смесь кипятят 5 мин. Отдельно в пробирке приготовляют смесь 2 мл 3%-ной перекиси водорода, 1 мл 3%-ной серной кислоты и 2 мл эфира. К этой смеси приливают около 1 мл охлажденной после кипячения пробы и основательно перемешивают взбалтыванием. В присутствии хрома слой эфира приобретает синюю окраску.
Определение с дифенилкарбазидом (шестивалентный хром). К 10 мл нейтрализованной пробы прибавляют 1 мл разбавленной (1:9) серной кислоты, несколько капель фосфорной кислоты и 0,5 мл 0,5%-ного раствора дифенилкарбазида в ацетоне. Появление через 10 мин красно-фиолетового окрашивания указывает на присутствие хрома (VI). Так можно обнаружить 0,05 мг/л хрома. (Мешающие влияния см. ниже.)
Колориметрическое определение с дифенилкарбазидом
28.3. Хроматы и бихроматы реагируют в кислой среде с дифенилкарбазидом с образованием растворимого соединения красно-фиолетового цвета, пригодного для колориметрирования. Шестивалентный хром определяют непосредственно (вариант А). Общее содержание хрома определяют после окисления персульфатом в кислой среде (вариант Б). Содержание трехвалентного хрома находят по разности результатов обоих определений. По описанной методике можно определить хром в неразбавленной пробе при содержании его от 0,05 до 1,0 мг в 1 л воды.
Мешающие влияния
28.4. Определению мешают присутствующие в высоких концентрациях (свыше 200 мг/л) ионы Hg2+ и  ; с дифенилкарбазидом вступают в реакцию и окрашивают раствор также ванадий и шестивалентный молибден, которые в воде обычно отсутствуют.
; с дифенилкарбазидом вступают в реакцию и окрашивают раствор также ванадий и шестивалентный молибден, которые в воде обычно отсутствуют.
Мешает определению также железо (1 мг/л), образующее с этим реактивом соединение, окрашивающее анализируемый раствор в желто-бурый цвет. Влияние железа можно частично устранить добавлением фосфорной кислоты, что предусмотрено в ходе определения. В присутствии больших количеств марганца при окислении персульфатом осаждается двуокись марганца. Ее в таких случаях отфильтровывают через стеклянную фильтрующую пластинку или через стеклянную вату.
При определении хрома (VI) на результаты может повлиять то обстоятельство, что хромат или бихромат могут окислять некоторые содержащиеся в пробе вещества в интервале времени между взятием пробы и ее анализом. В подобных случаях хром определяют "на месте" непосредственно после отбора пробы.
В нейтральных или щелочных водах раздельное определение хрома (III) и хрома (VI) затруднено тем, что при подкислении таких вод, если они (как это обычно бывает) содержат восстановители [соли железа (II), сульфиты, многие органические вещества и т.п.], происходит восстановление хрома (VI) до хрома (III).
В водах, окрашенных органическими веществами, непосредственно колориметрически определить хром (VI) трудно и тогда, когда эти воды имеют кислую реакцию. Во всех вышеприведенных случаях определяют только общее содержание хрома ("общий хром").
Аппаратура
28.5. Фотометр, светофильтр  .
.
.Кюветы толщиной 1 - 5 см или набор цилиндров Несслера емкостью 200 мл.
Реактивы
28.6. Едкий натр ч.д.а., 1 н. раствор: 40 г NaOH ч.д.а. растворяют в дистиллированной воде и доводят объем до 1 л.
Серная кислота ч.д.а., 1 н. раствор: 28 мл концентрированной H2SO4 ч.д.а. приливают к 500 мл дистиллированной воды и доводят до 1 л.
Серная кислота ч.д.а., разбавленная (1:1).
Фосфорная кислота ч.д.а., концентрированная, 85%-ная.
Дифенилкарбазид, 0,5%-ный раствор в ацетоне: 0,25 г дифенилкарбазида ч.д.а. растворяют в 50 мл ацетона ч.д.а. или в ацетоне, который предварительно кипятился с обратным холодильником в течение 3 ч с добавлением перманганата калия и был затем перегнан. Раствор хранят в коричневой склянке. Если раствор при стоянии окрасился, он для использования непригоден.
Персульфат аммония ч.д.а., 0,1%-ный раствор, свежеприготовленный.
Бихромат калия, стандартный раствор:
а) запасной: 2,8285 г K2Cr2O7 ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и дополняют объем при 20 °C до 1 л; 1 мл раствора содержит 1 мг Cr;
б) рабочий I: 25,0 мл запасного стандартного раствора разбавляют дистиллированной водой, доводя объем до 500 мл; 1 мл раствора содержит 0,050 мг Cr;
в) рабочий II: 20,0 мл рабочего раствора I разбавляют дистиллированной водой до 500 мл. Применяют каждый раз свежеприготовленный раствор; 1 мл раствора содержит 0,002 мг Cr.
Калибровочная кривая
28.7. В ряд мерных колб емкостью 100 мл отмеривают 0 - 1,0 - 2,0 - 5,0 - 10,0 - 15,0 - 20,0 - 30,0 - 40,0 - 50,0 мл рабочего стандартного раствора II, что после доведения объемов до 100 мл дает серию стандартных растворов с концентрациями хрома 0 - 0,02 - 0,04 - ... - 1,0 мг в 1 л. Затем проводят определение по варианту А. Из полученных значений оптической плотности вычитают величину оптической плотности холостого определения и полученные величины наносят на график против соответствующих концентраций хрома.
При визуальном определении в цилиндры Несслера отмеривают 0 - 2,0 - 4,0 - 6,0 - 8,0 - ... - 20,0 мл рабочего стандартного раствора, получая после разбавления водой до объема 100 мл серию стандартных растворов с концентрациями 0,0 - 0,04 - 0,08 - 0,12 - ... - 0,4 мг хрома в 1 л.
Для приготовления реактивов и разбавления пробы используют воду, дважды перегнанную в стеклянном приборе.
Ход определения
28.8. Вариант А (определение шестивалентного хрома). В мерную колбу емкостью 100 мл или в цилиндр Несслера отмеривают такой объем прозрачной пробы, чтобы в нем содержалось от 0,005 до 0,1 мг хрома. Пробу нейтрализуют необходимым количеством 1 н. раствора едкого натра или 1 н. раствора серной кислоты. Необходимое количество щелочи или кислоты устанавливается титрованием отдельной порции пробы. Затем приливают 1 мл серной кислоты (1:1), 0,3 мл фосфорной кислоты, доводят объем дистиллированной водой до 100 мл и перемешивают. Добавляют 2 мл раствора дифенилкарбазида и снова перемешивают. Через 5 - 10 мин после добавления дифенилкарбазида измеряют оптическую плотность или сравнивают окраску пробы со стандартными растворами, обработанными таким же способом. Из полученного значения оптической плотности пробы вычитают значение ее для холостого определения с дистиллированной водой и по калибровочной кривой находят содержание хрома.
28.9. Вариант Б (определение общего содержания хрома). Отмеривают 100 мл первоначальной неразбавленной, разбавленной или же сконцентрированной выпариванием пробы, содержащей в этом объеме 0,005 - 0,1 мг хрома, и нейтрализуют необходимым количеством 1 н. раствора едкого натра или 1 н. раствора серной кислоты. Это количество щелочи или кислоты определяют титрованием другой порции пробы. Затем прибавляют 0,3 мл 1 н. серной кислоты и 5 - 10 мл раствора персульфата аммония, после чего кипятят раствор 20 - 25 мин (при этом весь персульфат должен разложиться, так как следы его мешают последующему определению). Раствор выпаривают примерно до 50 мл, переносят в мерную колбу емкостью 100 мл или в цилиндр Несслера, после чего продолжают анализ по варианту А.
Расчет. Содержание хрома (VI) и общего хрома (x) в мг/л вычисляют по формуле

где c - содержание хрома, найденное по калибровочной кривой или сравнением с серией стандартных растворов, мг/л;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 0,005 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 |
Округление, мг/л | 0,005 | 0,01 | 0,02 | 0,05 |
Общие положения
29.1. В большинстве случаев цинк попадает в водоемы с промышленными сточными водами.
Цинк в воде встречается или в виде катионов цинка, или в форме цианидных и тартратных комплексов. Иногда цинк встречается в нерастворимых формах: в виде гидроокиси, карбоната, сульфида и т.д.
Для определения цинка в питьевой и поверхностной водах приводится колориметрический метод с дитизоном, который применяется при концентрациях цинка от сотых долей миллиграмма до целых миллиграммов в 1 л.
Пробы с малым содержанием цинка, если они не содержат цианидов, консервируют добавлением 1 мл концентрированной серной кислоты на 1 л воды. Для раздельного определения растворимой и нерастворимой форм цинка в пробе последнюю фильтруют сразу же после ее отбора. Нерастворимые соединения переводятся в раствор кипячением пробы с соляной кислотой (3 - 5 мл концентрированной соляной кислоты на 100 мл пробы). В исключительных случаях необходимо пробу выпарить досуха, остаток увлажнить соляной кислотой и снова выпарить.
Результаты определения приводят в миллиграммах цинка в 1 л воды.
Качественное определение
29.2. К 50 мл пробы добавляют несколько капель концентрированной соляной кислоты и после перемешивания с 20 мл ацетатного буферного раствора (получаемого смешением равных объемов 2 н. раствора ацетата натрия в 2 н. уксусной кислоты) добавляют 3 мл раствора тиосульфата натрия (50 г Na2S2O3·5H2O в 30 мл дистиллированной воды) и 3 мл раствора дитизона. После этого смесь тщательно встряхивают. (Приготовление раствора дитизона см. стр. 138). В присутствии цинка цвет органического слоя переходит в фиолетовый или красный.
Колориметрическое определение с дитизоном
29.3. Ионы цинка можно экстрагировать из водного раствора раствором дитизона в четыреххлористом углероде. Образующийся комплекс дитизона с цинком имеет красный цвет, и интенсивность окраски пропорциональна концентрации цинка. Цинк вступает в реакцию с дитизоном количественно в промежутке pH от 4 до 7. В этой среде с дитизоном реагируют также медь, кадмий, свинец, никель, кобальт, висмут, таллий, индий, ртуть, серебро, золото и палладий. Чтобы устранить мешающее влияние этих элементов, экстракцию проводят при pH = 5 с добавлением тиосульфата и цианида. В таких условиях с дитизоном помимо цинка реагирует только двухвалентное олово.
В обработанной пробе можно определить 0,005 - 0,030 мг цинка.
Мешающие влияния
29.4. Мешающее влияние большинства металлов устраняется обработкой пробы до экстрагирования, как это предусмотрено в ходе определения. В присутствии мешающих катионов в концентрациях, превышающих 0,5 мг в обработанной пробе, надо прибавить большее количество маскирующего реактива. Двухвалентное олово окисляется кипячением пробы с 0,5 мл 30%-ной перекиси водорода. Очень важно затем полностью удалить кипячением избыток перекиси водорода. После окисления пробу фильтруют и обрабатывают описанным ниже способом.
В пробе не должны присутствовать вещества, окисляющие дитизон с образованием соединений, окрашенных в коричневый цвет. Так реагируют, например, хлор, бром, йод, перекиси, трехвалентное железо. Свободные галогениды удаляют из раствора кипячением, железо осаждают в щелочной среде едким натром и отфильтровывают. После фильтрования пробу нейтрализуют и обрабатывают, как описано ниже.
Окрашенные вещества, экстрагируемые четыреххлористым углеродом, предварительно удаляют повторной экстракцией чистым растворителем. Последний экстракт должен быть бесцветным. Только после этого экстрагируют раствором дитизона.
Большие количества органических веществ могут помешать экстрагированию и вызвать помутнение четыреххлористого углерода. Такие пробы минерализуют выпариванием с 1 мл концентрированной серной кислоты, 2 мл концентрированной азотной кислоты и 0,5 мл 30%-ной перекиси водорода. Остаток после разложения растворяют в дистиллированной воде и обрабатывают, как описано ниже.
Высокая чувствительность дитизонового метода требует, чтобы при работе с этим реактивом соблюдались определенные правила, особенно необходима высокая степень чистоты применяемых реагентов, точное отмеривание реактивов и соблюдение одинакового хода определения при анализе пробы и в холостом опыте. Необходимо до минимума сократить расход реактивов, добавляемых при анализе, например вместо нейтрализации сильно кислых вод лучше пробу полностью выпарить, а остаток растворить в дистиллированной воде, иногда с прибавлением минимального количества кислоты.
Посуду для анализа следует тщательно промывать разбавленной азотной кислотой и раствором дитизона. Степень чистоты посуды, особенно делительных воронок, контролируют добавлением малого количества разбавленного раствора дитизона. Зеленая окраска последнего не должна изменяться.
Аппаратура
29.5. Фотометр, светофильтр  .
.
.Кюветы толщиной 0,5 - 1 см, снабженные крышками.
Реактивы
29.6. Дистиллированная вода. Для приготовления реактивов и для разбавления проб воду нужно перегнать в стеклянном приборе.
Ацетат натрия, 10%-ный раствор: 10 г CH3COONa·3H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Маскирующий раствор:
а) 0,15 г (NH4)2C2O4·H2O ч.д.а., 1,5 г KCN ч.д.а. и 24 г CH3COONa·3H2O ч.д.а. растворяют в 200 мл дистиллированной воды и добавляют 10 мл 2 н. раствора NH4OH (2,8 мл концентрированной NH4OH ч.д.а. в 10 мл раствора) и 70 мл 1 н. HCl (7 мл концентрированной HCl ч.д.а. в 70 мл раствора);
б) 120 г Na2S2O3·5H2O растворяют в 200 мл дистиллированной воды. Оба раствора соединяют и разбавляют водой до 1 л.
Четыреххлористый углерод ч.д.а. или очищенный перегонкой (см. стр. 107).
Дитизон, раствор для экстрагирования:
а) запасной: (приготовление см. стр. 138);
б) рабочий: к одной объемной части запасного раствора добавляют девять объемных частей четыреххлористого углерода. Раствор хранят в темной бутыли на холоду; он устойчив примерно одну неделю.
Промывной раствор (фосфат натрия + сульфид натрия): 6 г Na2HPO4·12H2O ч.д.а. растворяют в 100 мл дистиллированной воды, pH раствора доводится до 11 добавлением 5 н. раствора NaOH. После этого добавляют 10 мл 0,25 н. раствора сульфида натрия, полученного пропусканием сероводорода в 0,25 н. раствор едкого натра (10 г NaOH в 1 л дистиллированной воды). Величина pH этого раствора должна достичь значения 8. Раствор сульфида применяют всегда свежеприготовленным.
Сульфат цинка, стандартный раствор:
а) запасной: 1,00 г цинка ч.д.а. растворяют в 10 мл концентрированной азотной кислоты ч.д.а., добавляют 5 мл серной кислоты и выпаривают на водяной бане до появления белых паров H2SO4. Затем разбавляют дистиллированной водой до 1 л; 1 мл раствора содержит 1,000 мг цинка;
б) рабочий I: 25 мл запасного раствора смешивают с 5 мл концентрированной серной кислоты ч.д.а. и дистиллированной водой доливают до 1 л. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,025 мг Zn;
в) рабочий II: к 20,0 мл рабочего раствора I прибавляют 5 мл серной кислоты и доливают до 1 л дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,0005 мг цинка.
Калибровочная кривая
29.7. В делительные воронки отмеряют 0 - 10,0 - 20,0 - 30,0 - 40,0 - 50,0 - 60,0 мл рабочего стандартного раствора II, что соответствует после дополнения до 100 мл концентрациям 0 - 0,050 - 0,10 до 0,30 мг/л цинка. Стандартные растворы обрабатывают описанным ниже способом. Измеренные значения оптической плотности после вычитания из них оптической плотности холостого определения наносят на график против соответствующих концентраций цинка в мг/л.
Ход определения
29.8. В делительную воронку помещают 100 - 200 мл пробы, если надо предварительно разбавляя ее или упаривая, чтобы содержание цинка в этом объеме оказалось в пределах 0,005 - 0,03 мг, и прибавляют 1 мл концентрированной соляной кислоты ч.д.а., не содержащей цинка. Прибавляют раствор ацетата натрия до pH = 5, что проверяется индикаторной бумажкой, и 20 мл маскирующего раствора. После перемешивания экстрагируют цинк 50,0 мл рабочего раствора дитизона. Встряхивают до тех пор, пока окраска раствора не перестанет изменяться. Если экстракт окрашен в интенсивно фиолетовый или даже в красный цвет, экстракцию повторяют с меньшим объемом пробы. После хорошего разделения слоев сливают экстракт в другую делительную воронку, добавляют 10 мл промывного раствора, 15 мл дистиллированной воды и встряхивают до полного удаления дитизона из слоя четыреххлористого углерода, что отмечается исчезновением зеленой окраски. После полного разделения слоев сливают слой органического растворителя в кювету (конец делительной воронки прежде высушивают фильтровальной бумагой). Если раствор четыреххлористого углерода окажется мутным, фильтруют его через маленький сухой бумажный фильтр. Первые 5 мл фильтрата отбрасывают, а остальное собирают в кювету и измеряют оптическую плотность. Одновременно проводят холостой опыт со 100 мл дистиллированной воды, вводят поправку и по калибровочной кривой находят содержание цинка. (Если оптическая плотность холостого определения очень велика, надо измерить оптическую плотность экстракта пробы по отношению к экстракту холостого определения.)
Расчет. Содержание цинка (x) в мг/л вычисляют по формуле

где с - концентрация цинка по калибровочной кривой, мг/л;
V - объем пробы, взятой к определению, мл.
Округление результатов
Диапазон, мг/л | 0,010 - 0,050 | 0,050 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 |
Округление, мг/л | 0,002 | 0,005 | 0,01 | 0,02 |
Общие положения
30.1. Термином "анионоактивные синтетические моющие вещества" в настоящем руководстве названы поверхностно-активные соединения типа алкилсульфонатов и алкиларилсульфонатов. Их содержание в бытовых сточных водах может достигать 20 мг/л, а в промышленных сточных водах - даже граммов на 1 л. Присутствие синтетических моющих веществ в поверхностных и питьевых водах указывает на их загрязненность.
Колориметрическое экстракционное определение с метиленовой синей дает общее содержание анионоактивных синтетических моющих веществ. Биологически окисляющиеся синтетические моющие вещества требуют двукратного определения: перед биохимическим окислением и после него в аэробных условиях.
Пробы можно консервировать прибавлением 2 - 4 мл хлороформа на 1 л воды. Консервированной пробой нельзя пользоваться для описываемого ниже "Определения неокисляющихся анионоактивных синтетических моющих веществ."
Результаты выражают в миллиграммах синтетических моющих веществ в 1 л в пересчете на лаурилсульфонат натрия. Можно в качестве стандарта применять и другие моющие вещества, например, алкилмоносульфонат натрия R CH2SO3Na, где R = C14H29; при проведении арбитражных анализов нужно применять лаурилсульфонат натрия.
Колориметрическое определение с метиленовой синей
30.2. Анионоактивные синтетические моющие вещества образуют в водных растворах с метиленовой синей комплекс, экстрагируемый хлороформом. В щелочной среде возникает бесцветная лейкоформа, переходящая при изменении pH в окрашенную форму. В приведенной методике применяют экстракцию хлороформом из щелочной среды полученного окрашенного комплекса.
Интервал определяемых концентраций синтетических моющих веществ зависит от объемов взятой для анализа пробы и растворителя. Например, в 250 мл пробы воды можно определить 0,01 - 0,80 мг/л лаурилсульфоната натрия с точностью +/- 2%.
Ниже описаны два варианта хода анализа. В первом из них (вариант А) стандартным веществом является лаурилсульфонат натрия, во втором (вариант Б) - определяемое вещество.
Вариант А
Мешающие влияния
Определению мешают сульфиды, полисульфиды и тиосульфаты. Эти соединения удаляют окислением, осуществляемым добавлением 10 мл фосфатного буфера и 2 мл 20%-ной перекиси водорода на каждые 100 мл пробы. После 5 мин стояния можно приступить к определению. Мешающие влияния красителей, экстрагируемых в условиях определения, устраняют вычитанием оптической плотности хлороформного экстракта этих соединений.
Аппаратура
30.3. Делительные воронки емкостью 500 мл.
Фотометр, светофильтр  .
.
.Кюветы с толщиной слоя 5 или 2 см.
Реактивы
30.4. Двухзамещенный фосфат натрия, буферный раствор: 10 г Na2HPO4 растворяют в 900 мл дистиллированной воды и добавлением 1 н. раствора NaOH доводят pH до 10 и разбавляют до 1 л.
Метиленовый синий, нейтральный раствор: 0,35 г метиленовой синей растворяют в 1 л дистиллированной воды.
Метиленовый синий, кислый раствор: 0,35 г метиленовой синей растворяют в 500 мл дистиллированной воды, добавляют 6,5 мл концентрированной H2SO4 ч.д.а. и объем доводят до 1 л дистиллированной водой.
Хлороформ ч.д.а. или очищенный двукратной перегонкой.
Лаурилсульфонат натрия, стандартный раствор:
а) запасной: 1,000 г лаурилсульфоната натрия ч.д.а. растворяют в дистиллированной воде и после добавления 1 мл хлороформа доводят до 1 л; раствор надо сохранять в холодном месте; 2 - 3 недели он устойчив; 1 мл раствора содержит 1,00 мг лаурилсульфоната натрия;
б) рабочий: 10,0 мл запасного раствора разбавляют дистиллированной водой до 1000 мл; он всегда должен быть свежеприготовленным; 1 мл содержит 0,010 мг лаурилсульфоната натрия.
Калибровочная кривая
30.5. В ряд делительных воронок, содержащих 100 - 80 мл дистиллированной воды, отмеривают пипеткой 0 - 0,5 - 1,0 - 2,0 - 5,0 - 8,0 - 12,0 - 16,0 и 20,0 мл рабочего стандартного раствора, что отвечает 0,005 - 0,20 мг лаурилсульфата.
Эти растворы обрабатывают описанным ниже способом. Экстракты доводят до 50 мл хлороформом и измеряют оптическую плотность. Вычитают значение, полученное для холостой пробы, и наносят полученные величины на график против соответствующих концентраций лаурилсульфата. Для каждого нового препарата метиленовой синей всегда следует строить новую калибровочную кривую.
Примечание. Все хлороформные экстракты собирают после проведения определений и сохраняют. Затем отгоняют хлороформ и очищают его двукратной перегонкой. Применяемую посуду ополаскивают разбавленной (1:4) азотной кислотой и дистиллированной водой.
Ход определения
30.6. Если предполагают, что концентрация синтетических моющих веществ ниже 0,8 мг/л, то отмеривают в одну делительную воронку 250 мл пробы (при ожидаемом содержании 0,8 - 2 мг/л - 100 мл, при содержании 2 - 4 мг/л - 50 мл и т.д.). Если взятый объем пробы меньше 100 мл, его доводят до 100 мл дистиллированной водой. На каждые 100 мл добавляют 10 мл фосфатного буфера (если прежде не производилось подкисление в целях устранения мешающих веществ), перемешивают и прибавляют 5 мл нейтрального раствора метиленовой синей и 15 мл хлороформа.
Во вторую делительную воронку наливают 100 мл дистиллированной воды и 5 мл кислого раствора метиленовой синей. Смесь в первой делительной воронке осторожно взбалтывают 2 мин. После расслоения спускают хлороформный слой во вторую делительную воронку. В первую воронку снова добавляют 5 мл хлороформа, взбалтывают 2 мин, хлороформный слой вновь спускают во вторую делительную воронку. Этот процесс повторяют еще один раз. Содержимое второй делительной воронки взбалтывают 2 мин и после расслоения сливают прозрачный хлороформный слой в мерную колбу емкостью 50 мл через воронку с кусочком ваты для отделения возможно образовавшейся мути. Во вторую делительную воронку добавляют 5 мл хлороформа, взбалтывают 2 мин и слой растворителя также сливают в мерную колбу. Этот процесс повторяют еще один раз. Хлороформный экстракт разбавляют хлороформом до 50 мл и затем колориметрируют. Содержание анионоактивного вещества находят по калибровочной кривой.
Синяя окраска экстрактов устойчива, следовательно, ее интенсивность можно измерять сразу у нескольких проб.
Если измеряемая величина оптической плотности превышает максимальное значение калибровочной кривой, хлороформный экстракт соответственно разбавляют.
Для каждого ряда проб производят холостое определение с дистиллированной водой. Если хлороформный экстракт надо разбавить, то необходимо определить величину оптической плотности холостого определения при одинаковом разбавлении. Аналогично поступают в том случае, если экстрагирование производят в объемах, меньших, чем указано в инструкциях.
Расчет. Содержание анионоактивных синтетических моющих веществ (лаурилсульфонат натрия) (x) в мг/л вычисляют по формуле

где V1 - объем хлороформного экстракта, мл;
c - концентрация синтетических моющих средств, определенная по калибровочной кривой, мг/л;
V2 - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 0,005 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 |
Округление, мг/л | 0,005 | 0,01 | 0,02 | 0,05 |
Вариант Б
Мешающие влияния
Применяют двойное экстрагирование, которое устраняет мешающее действие хлоридов, нитратов, роданидов и белков. Определению мешают сульфиды, полисульфиды, тиосульфаты, восстанавливающие метиленовую синюю, но их влияние может быть исключено предварительным окислением перекисью водорода.
Эти соединения удаляют окислением, добавляя для этого 10 мл фосфатного буфера и 2 мл 20%-ной перекиси водорода на каждые 100 мл пробы. После 5 мин стояния можно приступить к определению.
Аппаратура, см. стр. 154.
30.7. Реактивы, см. стр. 155, а также стандартный раствор определяемого моющего вещества: растворяют 0,100 г определяемого моющего вещества в дистиллированной воде и разбавляют раствор до 1 л. Отобрав 10 мл раствора, разбавляют его дистиллированной водой до 100 мл. В 1 мл полученного разбавленного раствора содержится 0,01 мг моющего вещества.
Калибровочная кривая
30.8. Отбирают порции: 2; 5; 10; 15; ... 30 мл стандартного раствора моющего вещества, разбавляют каждую порцию дистиллированной водой до 100 мл и продолжают, как указано в ходе определения.
Ход определения
30.9. Отбирают объем анализируемой сточной воды, чтобы в нем содержалось от 20 до 300 мкг определяемого моющего вещества. Если в 100 мл сточной воды присутствует менее 20 мкг синтетического моющего вещества, отбирают соответственно больший ее объем и упаривают до объема менее 100 мл.
Отобранную порцию пробы переносят в делительную воронку емкостью 200 - 250 мл, разбавляют дистиллированной водой до 100 мл, прибавляют 10 мл фосфатного буферного раствора, 5 мл нейтрального раствора метиленовой синей и 15 мл хлороформа.
Осторожно взбалтывают в течение 1 мин и дают постоять 1 мин для расслоения жидкости. Затем сливают слой хлороформа в другую делительную воронку такой же емкости, в которую предварительно наливают 110 мл дистиллированной воды и 5 мл кислого раствора метиленовой синей.
Содержимое второй воронки взбалтывают так же, как и содержимое первой, дают жидкости расслоиться и сливают нижний хлороформный слой через маленькую воронку, в которую предварительно помещают тампон обыкновенной ваты, пропитанный хлороформом, в мерную колбу емкостью 50 мл.
Затем в первую воронку наливают еще 10 мл хлороформа и повторяют описанные выше операции. Экстракцию проводят еще один раз порцией хлороформа в 10 мл и один раз 5 мл. Всего в мерной колбе должно собраться около 40 мл хлороформных экстрактов.
Доливают содержимое колбы до метки хлороформом и перемешивают. Окрашенный хлороформный раствор переносят в кювету с расстоянием между стенками, равным 3 см, и измеряют его оптическую плотность, поместив во вторую кювету раствор "холостого опыта", для которого берут 100 мл дистиллированной воды.
Определение неокисляющихся биохимически анионоактивных
синтетических моющих веществ
30.10. Содержание неокисляющихся синтетических моющих веществ определяют в пробе после пятидневной инкубации при температуре 20 °C в темноте в присутствии кислорода.
Аппаратура
30.11. Термостат (20 °C), остальная аппаратура такая же, как и для колориметрического определения общего содержания синтетических моющих веществ (см. стр. 154).
Реактивы
30.12. Разбавляющая вода с добавками, приготовление см. стр. 82.
Ход определения
30.13. Определяют общее содержание синтетических моющих веществ колориметрическим методом с метиленовой синей, как описано выше. В чистую стеклянную склянку емкостью около 500 мл, снабженную притертой пробкой, вводят 200 мл пробы с известным уже количеством синтетических моющих средств и доводят до 300 мл разбавляющей водой. Концентрация синтетических моющих средств в исследуемой пробе не должна превышать 2 мг/л. Склянку закрывают стеклянной пробкой и помещают в термостат на 5 дней. Содержимое склянки взбалтывают 2 - 3 раза в сутки. Через 5 суток синтетические моющие вещества определяют в пробе с инкубацией колориметрическим методом с метиленовой синей.
Расчет. Содержание определяемых веществ (x) в мг/л после пятидневной инкубации вычисляют по формуле, приведенной на стр. 156.
Результат соответствует приблизительно количеству биохимически неокисляющихся синтетических моющих средств. Содержание окисляющихся синтетических моющих веществ вычисляют по разности между общим содержанием синтетических моющих веществ, найденных перед началом инкубации, и содержанием их, найденным на пятый день.
Округление результатов. Результаты округляют так же, как и при колориметрическом определении общего содержания.
Общие положения
31.1. Бораты содержатся в небольших количествах в поверхностных водах и являются их естественной составной частью. Содержание боратов возрастает в промышленных и бытовых сточных водах, образующихся при применении или производстве различных стиральных порошков.
Пробу следует отбирать в полиэтиленовые бутылки или в бутылки из стекла, не содержащего бора.
Для определения боратов в концентрациях от 2 до 20 мг/л приводится колориметрический метод с карминовой кислотой; для определения более высоких концентраций предлагается объемный метод. При анализе первым методом концентрацию пробы можно изменить предварительным ее выпариванием или разбавлением.
Результаты выражаются в миллиграммах содержания метаборат-ионов в 1 л.
Качественное определение
31.2. К 100 мл пробы прибавляют 5%-ный раствор карбоната натрия до слабой щелочной реакции. Потом прибавляют несколько капель спиртового раствора экстракта куркумы и смесь выпаривают досуха. Появление красновато-коричневой окраски является доказательством присутствия боратов. Чувствительность при анализе 100 мл пробы составляет примерно 0,02 мг  в 1 л воды.
в 1 л воды.
Колориметрическое определение с карминовой кислотой
31.3. Раствор карминовой кислоты в серной кислоте в присутствии боратов меняет окраску из красной в синюю. В присутствии небольшого количества органических веществ, не разбавляя пробу (вариант А), можно определить от 2 до 20 мг  в 1 л воды с ошибкой +/- 0,5 мг. Для определения более низких концентраций, а также в присутствии большого количества органических веществ предлагается вариант Б.
в 1 л воды с ошибкой +/- 0,5 мг. Для определения более низких концентраций, а также в присутствии большого количества органических веществ предлагается вариант Б.
Мешающие влияния
31.4. Ионы, содержащиеся обычно в водах, не мешают определению.
Аппаратура
31.5. Платиновая чашка.
Центрифуга.
Фотометр, светофильтр 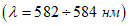 .
.
.Кюветы толщиной не менее 1 см; цилиндры Несслера емкостью 50 мл.
Муфельная или тигельная печь (500 - 550 °C).
Реактивы
31.6. Все реактивы приготовляют и сохраняют в склянках из химически устойчивого безборатного стекла.
Едкий натр, примерно 1 н. раствор: 40 г NaOH растворяют в дистиллированной воде и разбавляют до 1 л.
Соляная кислота ч.д.а., концентрированная.
Серная кислота ч.д.а., концентрированная.
Карминовая кислота, 0,05%-ный раствор: 0,05 г карминовой кислоты растворяют в 100 мл концентрированной серной кислоты.
Соляная кислота ч.д.а., разбавленная (1:1).
Борная кислота, стандартный раствор:
а) запасной: 1,4446 г ортоборной кислоты H3BO3 ч.д.а., высушенной в эксикаторе, растворяют в дистиллированной воде и объем раствора доводят до 1 л; 1 мл содержит 1,00 мг  ;
;
б) рабочий: 25,0 мл запасного раствора разбавляют до 1 л дистиллированной водой. Пользуются всегда свежеприготовленным раствором; 1 мл раствора содержит 0,025 мг 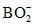 .
.
Калибровочная кривая
31.7. Вследствие нестойкости карминовой кислоты калибровочную кривую надо строить заново для каждого ряда определений. В мерные колбы емкостью в 50 мл наливают 0 - 4,0 - 10,0 - 20,0 - 30,0 - 40,0 мл стандартного раствора и разбавляют дистиллированной водой до метки. Таким образом, получается ряд стандартных растворов, содержащих 0 - 2,0 - 5,0 - 10,0 - 15,0 - 20,0 мг  в 1 л, которые обрабатывают по варианту А.
в 1 л, которые обрабатывают по варианту А.
Стандартные растворы для определения в цилиндрах Несслера приготавливают таким же способом.
Ход определения
31.8. Вариант А (проба содержит небольшое количество органических веществ). К 2 мл пробы, содержащей от 2 до 20 мг  в 1 л, прибавляют в небольшой колбе две капли концентрированной соляной кислоты. Потом приливают 10 мл концентрированной серной кислоты, смесь тщательно перемешивают и охлаждают. После охлаждения прибавляют 10 мл раствора карминовой кислоты, смесь снова перемешивают и оставляют в покое в течение 45 мин. Измеряют оптическую плотность. Таким же образом определяют оптическую плотность холостого определения с дистиллированной водой. При определении в цилиндрах Несслера окраску полученного раствора сравнивают с рядом стандартов.
в 1 л, прибавляют в небольшой колбе две капли концентрированной соляной кислоты. Потом приливают 10 мл концентрированной серной кислоты, смесь тщательно перемешивают и охлаждают. После охлаждения прибавляют 10 мл раствора карминовой кислоты, смесь снова перемешивают и оставляют в покое в течение 45 мин. Измеряют оптическую плотность. Таким же образом определяют оптическую плотность холостого определения с дистиллированной водой. При определении в цилиндрах Несслера окраску полученного раствора сравнивают с рядом стандартов.
Вариант Б (проба с содержанием большого количества органических веществ). К 5,0 мл раствора прибавляют 1 н. раствор едкого натра до явно щелочной реакции, выпаривают в платиновой чашке на водяной бане досуха и прокаливают остаток примерно при 500 - 550 °C. При охлаждении прибавляют к остатку 5,0 мл разбавленной соляной кислоты и растирают содержимое чашки стеклянной палочкой с резиновой трубкой на конце. Смесь переносят в центрифужную пробирку и центрифугируют до прояснения. Отбирают 2 мл прозрачного раствора в небольшую колбу и далее продолжают так же, как в варианте А. Таким же способом проводят холостой опыт с дистиллированной водой.
При анализе предварительно сконцентрированных проб, содержащих менее 2 мг/л  , ход определения такой же. Объем пробы, взятой на определение, должен содержать не менее 0,010 мг
, ход определения такой же. Объем пробы, взятой на определение, должен содержать не менее 0,010 мг  .
.
Расчет. Содержание борат-ионов в мг/л, определенное в присутствии небольших (x) или больших (y) количеств органических веществ, вычисляют по формулам:

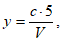
где c - концентрация боратов, полученная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 2,0 - 5,0 | 5,0 - 10,0 | 10 - 20 | 20 - 50 |
Округление, мг/л | 0,2 | 0,5 | 1 | 2 |
Более высокие и более низкие результаты округляются по приведенной схеме.
Колориметрический метод с куркумином
31.9. Если проба воды, содержащая бор, подкислена и выпарена в присутствии куркумина, образуется красно-окрашенный продукт, называемый розоцианин. Розоцианин легко растворим, и красный цвет его растворов сравнивается со стандартами визуально или фотометрически.
Мешающие влияния
31.10. Ионы, обычно встречающиеся в воде, не мешают при этом методе, за исключением нитратов в концентрации свыше 20 мг/л. Минимум открываемой концентрации: 0,2 мг  в 1 л воды.
в 1 л воды.
Аппаратура
31.11. Фотометр, светофильтр  .
.
.Чашка для выпаривания объемом 45 - 150 мл, из стекла, платины или другого материала, удобного для использования.
Водяная баня (55 +/- 2 °C).
Мерные колбы емкостью 25 мл, со стеклянными пробками.
Реактивы
31.12. Борная кислота, стандартный раствор (приготовление см. стр. 159).
Раствор куркумина: растворяют 0,040 г куркумина и 5,0 г щавелевой кислоты в 80 мл 95%-ного этилового спирта. Добавляют 4,2 мл концентрированной HCl и доводят раствор до 100 мл этиловым спиртом в 100 мл мерной колбе (вместо этилового спирта может быть использован 45%-ный изопропиловый спирт). Этот реактив будет устойчивым несколько дней, если хранить в холодильнике.
Этиловый спирт, 95%-ный.
Калибровочная кривая
31.13. Отмеряют пипеткой 0,00 (пустая), 0,25; 0,50; 0,75 и 1,00 мг рабочего раствора борной кислоты в чашки для выпаривания одного типа, формы и размера. Добавляют 4,0 мл реактива куркумина в каждую и тихонько встряхивают чашку, чтобы перемешать тщательно содержимое.
Ход определения
31.14. Предостережения! Для успешного определения необходим строгий контроль за изменением объема и концентрации реактивов, продолжительностью и температурой высушивания. Чашки для выпаривания должны быть одинаковые по форме, размеру и составу покрытия (глазури). Увеличение продолжительности выпаривания вызывает увеличение интенсивности окраски.
Для вод, содержащих 0,10 - 1,00 мг/л бора, берут 1,00 мл пробы. Для вод, содержащих больше, чем 1,00 мг/л бора, применяют соответствующее разбавление дистиллированной водой, свободной от бора, так чтобы 1,00 мл содержал приблизительно 0,50 мг бора.
Отбирают пипеткой 1,00 мл пробы или раствора в чашку для выпаривания.
Чашки помещают на водяную баню при температуре 55 +/- 2 °C и выпаривают содержимое до удаления влаги. Как только содержимое чашек станет сухим и исчезнет запах HCl, их снимают с водяной бани. Затем чашки охлаждают при комнатной температуре, добавляют 10 мл 95%-ного этилового спирта в каждую чашку, помешивая осторожно полиэтиленовым стержнем, чтобы завершить растворение красно-окрашенного продукта.
Смывают содержимое каждой чашки 95%-ным этиловым спиртом в мерную колбу емкостью 25 мл.
Тщательно перемешивают, переворачивая колбу, и определяют оптическую плотность с оранжевым светофильтром, фотометрируют не позже чем через 1 ч после высушивания пробы, и определяют содержание бора по калибровочной кривой.
Для низких концентраций боратов (0,02 - 0,05 мг/л) фотометрический метод может быть заменен визуальным. Для этого разбавляют рабочий раствор боратов, так чтобы 1 мл соответствовал 0,20 мг бора. Отбирают пипеткой растворы, содержащие 0,00, 0,05, 0,10, 0,15 и 0,20 мг бора, в чашки для выпаривания, как указано выше. Одновременно 1,00 мл или меньшее количество пробы (0,02 - 0,05 мг  ) помещают в такую же чашку и обрабатывают, как описано выше. Сравнение окрасок пробы и стандартных растворов производят не позже чем через 1 ч после растворения красного осадка в спирте.
) помещают в такую же чашку и обрабатывают, как описано выше. Сравнение окрасок пробы и стандартных растворов производят не позже чем через 1 ч после растворения красного осадка в спирте.
Расчет. Содержание  (x) в мг/л вычисляют по формуле
(x) в мг/л вычисляют по формуле

где a2 - оптическая плотность стандартного раствора;
а1 - оптическая плотность пробы;
c - содержание  в стандартном растворе, мг/л;
в стандартном растворе, мг/л;
V - объем пробы, мл.
Общие положения
32.1. Двуокись хлора - одно из новых средств, применяемых при улучшении качества воды для ее обеззараживания и для устранения различных нежелательных ее свойств, если для этого нельзя применить хлор. Одну двуокись хлора можно определять любым методом, применяемым для определения хлора.
Для определения двуокиси хлора в присутствии хлора и хлоритов, что представляет собой наиболее часто применяемый вид контроля в области водоснабжения, приводится ортотолидиновый метод, предназначенный для концентрации ClO2 от 0,05 мг/л.
Относительно точные результаты получаются при строгом соблюдении условий проведения анализа, особенно интервалов времени между отдельными операциями.
Для определения двуокиси хлора в присутствии хлора и хлоритов в растворах реагентов для обеззараживания воды и в стандартных растворах приводится йодометрический метод, пригодный при содержании примерно от 2 мг/л ClO2. Одновременно определяются хлор и хлориты.
Пробы нельзя консервировать; анализировать их необходимо тотчас после взятия.
Результаты определения выражаются в миллиграммах ClO2 в 1 л воды.
Колориметрическое определение с ортотолидином
32.2. Двуокись хлора дает с ортотолидином при pH = 1,9 желтую окраску, интенсивность которой пропорциональна содержанию ClO2. Чувствительность определения - 0,05 мг ClO2 в 1 л воды.
Мешающие влияния
32.3. Определению мешает свободный хлор, который удаляют щавелевой кислотой (вариант А) или малоновой кислотой (вариант Б). При значительном избытке хлора ни один из этих методов применить нельзя. Определению по варианту А мешает присутствие кальция в количествах, превышающих 100 мг/л, так как он осаждается щавелевой кислотой. В таких случаях применяют вариант Б. Хлориты реагируют подобно двуокиси хлора.
Аппаратура
32.4. Фотометр, светофильтр  .
.
.Кюветы длиной не менее 2 см.
Реактивы
32.5. Ортотолидин, 0,1%-ный раствор: 1 г ортотолидина ч.д.а. с 5 мл разбавленной (1:4) HCl ч.д.а. растирают в ступке до состояния жидкой пасты. Добавляют 200 мл дистиллированной воды и, когда ортотолидин растворится, переливают смесь в мерную колбу емкостью 1 л и разбавляют дистиллированной водой примерно до 450 мл. Добавляют 500 мл разбавленной (1:4) HCl ч.д.а. и доводят объем до метки дистиллированной водой. Раствор хранят в темной склянке.
Серная кислота ч.д.а., разбавленная (1:9).
Щавелевая кислота, насыщенный раствор (для варианта А): растворяют 10 г (COOH)2·2H2O ч.д.а. в 100 мл дистиллированной воды.
Малоновая кислота, 1%-ный раствор (для варианта Б): 1 г малоновой кислоты растворяют в дистиллированной воде и разбавляют раствор до 100 мл.
Двуокись хлора, стандартный раствор.
Для обычного производственного контроля можно ограничиться приготовлением стандартного раствора основного (см. ниже). Этот стандартный раствор помимо небольшого количества хлора может содержать и некоторое количество хлоритов. Для точной аналитической работы, равно как и в случаях, когда требуется стандартный раствор чистой двуокиси хлора без хлоритов, следует готовить раствор II.
Основной раствор I. В дистиллированной воде растворяют 1,34 г технического хлорита натрия NaClO2 (продается примерно 80%-ный препарат), 0,55 г гипохлорита натрия NaClO (берут раствор, в котором содержание гипохлорита было определено йодометрическим титрованием) и 0,72 г серной кислоты ч.д.а., т.е. 38 мл разбавленной (1:99) H2SO4, и разбавляют объем смеси до 1 л; в 1 мл приготовленного раствора содержится около 0,3 - 0,6 мг ClO2. Раствор хранят в темной склянке в холодном месте. Он сохраняет устойчивость около двух недель. В нем следует часто проверять содержание ClO2 и Cl2. Если в растворе окажется большое количество свободного хлора, то приготовляют свежий раствор. Содержание ClO2, Cl2 и 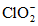 в стандартном растворе определяют йодометрическим методом (см. ниже).
в стандартном растворе определяют йодометрическим методом (см. ниже).
Основной раствор II. Растворяют 5 г технического хлорита натрия в 400 мл дистиллированной воды. Раствор наливают в реакционную колбу емкостью 1000 мл со шлифом. В колбу введена трубка, подводящая воздух и заканчивающаяся у дна стеклянным фильтром. Кроме того, в нее вводят капельную воронку с подводящей трубкой (она доходит до дна колбы) и отводную трубку, идущую к одной из соединенных между собой промывалок емкостью 500 или 1000 мл, снабженных стеклянными фильтрующими пластинками. В этом приборе трубки соединяют встык, используя короткие муфты из поливинилхлорида.
Первую промывалку примерно до одной трети объема заполняют твердым хлоритом натрия, предназначенным для поглощения свободного хлора. Во вторую промывалку наливают 750 - 800 мл дистиллированной воды для улавливания двуокиси хлора.
Собрав установку, пропускают в реакционную колбу медленный ток воздуха, проходящий затем через всю систему. Через капельную воронку вводят в 3 - 4 приема с интервалами в 3 - 5 мин всего 20 мл 10%-ного раствора серной кислоты. После этого продолжают пропускать воздух через установку еще не менее 30 мин.
Следует заботиться о безукоризненном соединении частей установки, контролировать места возможных повреждений, чтобы не допустить утечки газообразной ClO2 из аппаратуры. При соприкосновении этого газа, например, с пылью воздуха может произойти взрыв.
Приготовленный раствор содержит около 0,2 - 0,4 мг ClO2 в 1 мл (с примесью 1 - 2% свободного хлора).
Рабочий раствор. Его приготовляют разбавлением стандартного раствора. Содержание ClO2 в нем контролируют йодометрически. Каждый раз приготовляют свежий раствор. Хранят в темной склянке в холодном месте. Раствор очень неустойчив. Целесообразно приготовлять растворы, содержащие 0,01 мг ClO2 в 1 мл.
Калибровочная кривая
32.6. В ряд колб отмеривают 0 - 0,5 - 1,0 - 2,0 - 4,0 - 6,0 - 8,0 - 10,0 мл рабочего стандартного раствора (или иные соответствующие количества, если в 1 мл этого раствора содержится не точно 0,01 мг ClO2) с таким расчетом, чтобы после дополнения содержимого колб дистиллированной водой до 100 мл получить серию стандартных растворов с концентрациями: 0 - 0,05 - 0,1 - ... - 1,0 мг ClO2 в 1 л. Эти стандартные растворы обрабатывают согласно описанному ниже варианту А или Б. Определяют оптическую плотность, вычитают значение ее для холостого определения и строят график зависимости оптической плотности от концентраций ClO2 в мг/л.
Ход определения
32.7. Вариант А (с использованием щавелевой кислоты). К 100 мл прозрачной пробы (или к меньшему ее объему, дополненному до 100 мл дистиллированной водой) добавляют 0,5 мл разбавленной серной кислоты, 5 мл насыщенного раствора щавелевой кислоты и по истечении 10 мин приливают 1 мл раствора ортотолидина. Через 5 - 10 мин измеряют оптическую плотность, вводят поправку на холостое определение с дистиллированной водой и по калибровочной кривой определяют содержание ClO2.
Вариант Б (с использованием малоновой кислоты). К 100 мл прозрачной пробы (или к меньшему ее объему, дополненному до 100 мл дистиллированной водой) добавляют 2 мл раствора малоновой кислоты, а спустя 3 мин 1 мл раствора ортотолидина. Выждав точно еще 3 мин, измеряют оптическую плотность, вычитают ее значение для холостого определения с дистиллированной водой и по калибровочной кривой находят содержание ClO2.
Расчет. Содержание двуокиси хлора (x) в мг/л вычисляют по формуле

где c - содержание ClO2, найденное по калибровочной кривой для примененного варианта А или Б, мг/л;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 0,05 - 1,00 | 1,0 - 2,00 | 2,0 - 5,0 | 5,0 - 10,0 |
Округление, мг/л | 0,05 | 0,1 | 0,2 | 0,5 |
Йодометрическое определение двуокиси хлора
с одновременным определением хлора и хлоритов
32.8. Определение основано на трех йодометрических титрованиях. В первом из них, проводимом в нейтральной среде, расход титрованного раствора тиосульфата эквивалентен 1/5 суммы ClO2 + Cl2, во втором титровании, проводимом в кислой среде, титруется остаток ClO2 и NaClO2, т.е. 4/5 общего количества ClO2 и NaClO2. Третье титрование проводят в кислой среде после удаления из пробы ClO2 и Cl2 при pH = 7,0. Расход титрованного раствора тиосульфата эквивалентен содержанию хлорита.
Мешающие влияния
32.9. Результаты первого титрования могут изменяться при изменении pH среды. Следует титровать при pH более 6,0. Этого достигают добавлением к пробе буферного раствора.
Аппаратура
32.10. Стальной баллон со сжатым воздухом или азотом.
Сосуд для отдувки газа емкостью 500 мл.
Реактивы
32.11. Фосфатный буферный раствор pH = 7: растворяют 72,44 г Na2HPO4·2H2O ч.д.а. и 35,40 г KH2PO4 ч.д.а. в дистиллированной воде и доводят объем до 1 л.
Йодид калия ч.д.а.
Тиосульфат натрия, 0,05 н. титрованный раствор: приготовление см. стр. 76. Поправку к титру раствора определяют способом, описанным в разделе 12, используя 0,05 н. раствор бихромата калия.
Крахмал, 0,5%-ный раствор: приготовление см. стр. 63.
Серная кислота, приблизительно 1 н. раствор: осторожно вливают 27 мл концентрированной H2SO4 ч.д.а. в дистиллированную воду и дополняют объем до 1 л.
Ход определения
32.12. К 75 мл дистиллированной воды в колбе для титрования добавляют 5 мл фосфатного буферного раствора, 2 г йодида калия и осторожно приливают такой отмеренный объем пробы, чтобы в нем содержалось от 2 до 100 мг ClO2. Перемешав смесь, титруют ее 0,05 н. раствором тиосульфата по крахмалу до исчезновения окраски; израсходованный на это титрование объем a мл раствора записывают.
После первого титрования к раствору добавляют 50 мл 1 н. раствора серной кислоты (или же соответственно больший объем, если объем титруемой смеси превышает 100 мл) и, перемешав содержимое колбы, помещают ее на 10 мин в темное место. Затем титруют 0,05 н. раствором тиосульфата по крахмалу до исчезновения окраски. Записывают расход раствора тиосульфата b мл на второе титрование.
К новым 100 мл пробы, отмеренным в сосуд для отдувки газа (или же к иному подходящему объему - в зависимости от предполагаемого содержания хлорита), добавляют 5 мл фосфатного буферного раствора (и соответственно больше, если объем титруемой смеси превышает 100 мл) и продувают смесь в течение 20 - 30 мин воздухом или азотом для удаления ClO2 и Cl2. В целях безопасности вытесненные газы собирают в другой сосуд, до половины наполненный разбавленным раствором едкого натра. Затем смесь в первом сосуде подкисляют, приливая 50 мл 1 н. раствора серной кислоты на каждые 100 мл ее объема, добавляют 2 г йодида калия и, перемешав, титруют 0,05 н. раствором тиосульфата по крахмалу до исчезновения окраски. Записывают расход раствора тиосульфата c в миллилитрах.
Расчет. Содержание двуокиси хлора ClO2 (x), хлора Cl2 (y), хлоритов  (z) и хлорита натрия NaClO2 (m), все в мг/л, вычисляют по формулам:
(z) и хлорита натрия NaClO2 (m), все в мг/л, вычисляют по формулам:



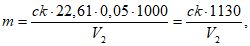
где a - расход 0,05 н. раствора тиосульфата на первое титрование, мл;
b - расход 0,05 н. раствора тиосульфата на второе титрование, мл;
c - расход 0,05 н. раствора тиосульфата на третье титрование, мл;
k - поправка для приведения нормальности раствора тиосульфата к точно 0,05 н.;
V1 - объем пробы, взятой для первого и второго титрования, мл;
V2 - объем пробы, взятой для третьего титрования, мл;
16,83 - эквивалент ClO2;
34,45 - эквивалент Cl2;
22,61 - эквивалент NaClO2.
Округление результатов
Диапазон, мг/л | 2,0 - 5,0 | 5,0 - 10,0 | 10 - 20 | 20 - 50 |
Округление, мг/л | 0,2 | 0,5 | 1 | 2 |
Общие положения
33.1. йод находится в природных водах преимущественно в виде йодидов. Нахождение его в подземных водах обусловлено составом подземных слоев. В поверхностных водах йод встречается редко. В эти воды он попадает из стоков. Соединения йода содержат сточные воды некоторых специальных отраслей химической и фармацевтической промышленности.
Для определения малых количеств йодидов (порядка 0,001 - 0,01 мг/л) в питьевой и поверхностной водах приводится каталитический метод. Для определения больших концентраций применяется йодометрический объемный метод. Пробы не консервируют.
Результаты определений даются в мг йодид-ионов на 1 л воды.
Каталитический метод с колориметрическим окончанием
33.2. Йодиды являются катализатором восстановления четырехвалентного церия мышьяковистой кислотой. Каталитическое действие пропорционально концентрации йодидов, если соблюдаются постоянные условия реакции, т.е. кислотность раствора, температура, продолжительность реакции и концентрации реагирующих веществ. Через определенное время прохождения реакции определяют концентрации четырехвалентного церия. Избыточным количеством четырехвалентного церия в варианте А окисляют добавленную соль двухвалентного железа, и образующееся трехвалентное железо определяют колориметрически с роданидом. В варианте Б избыток ионов четырехвалентного церия определяют непосредственно по их желтой окраске.
Пользуясь вариантом А, можно без изменения объема пробы определить 0,02 - 0,08 мг йодид-ионов в 1 л. Диапазон концентраций йодидов, которых можно определить по варианту Б, зависит от выбранных условий реакции. Если проба содержит йодиды в малых концентрациях, ее можно сконцентрировать упариванием при pH больше 7; при слишком высоких концентрациях йодидов в пробе ее разбавляют дистиллированной водой, свободной от йодидов.
Мешающие влияния
33.3. Определению мешают вещества, обладающие таким же каталитическим действием, как и йодиды. К ним относятся соли осмия и рения, частично бромиды и хлориды. Определению мешают также ионы, образующие с йодид-ионами нерастворимые соединения (ионы ртути, серебра, свинца). Мешают также цианиды, роданиды и все другие соединения, восстанавливающие четырехвалентный церий.
Влияние хлоридов снижается добавлением избыточного количества хлорида натрия, как это описано в ходе анализа. Железо мешает определению, если концентрация его превышает 10 мг/л. Железо удаляют пропусканием пробы через катионит в Na-форме или осаждением в виде гидроокиси.
Мешающее влияние органических и других веществ, способных окисляться солью четырехвалентного церия, устраняют минерализацией в щелочной среде. Отмеренный объем пробы почти полностью выпаривают в чашке из химически устойчивого стекла. Затем прибавляют 2 мл 2 н. раствора едкого кали (112 г KOH растворяют в дистиллированной воде и разбавляют до 1 л), 1 мл 10%-ного раствора сульфата цинка и по мере надобности несколько капель 10%-ного раствора перхлората калия. Смесь выпаривают досуха и остаток прокаливают при 600 °C. По охлаждении остаток в чашке растворяют в дистиллированной воде, раствор фильтруют, обмывая чашку и фильтр дистиллированной водой, и разбавляют фильтрат до определенного объема.
Аппаратура
33.4. Водяная баня, 30 +/- 0,5 °C (при анализе способом А).
Тонкостенные пробирки из химически устойчивого стекла емкостью около 20 мл.
Водяная баня со льдом.
Фотометр, светофильтр  для варианта А; светофильтр
для варианта А; светофильтр  для варианта Б.
для варианта Б.
для варианта А; светофильтр для варианта Б.Реактивы
33.5. Дистиллированная вода, применяемая для разбавления проб и для приготовления реактивов, не должна содержать более 0,0003 мг йодид-ионов в 1 л. От следов йодидов избавляются фильтрованием воды через слой анионита в OH-форме.
Хлорид натрия, 20%-ный раствор: 200 г NaCl ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л. Рекомендуется применять соль, перекристаллизованную из смеси спирт-вода.
Мышьяковистая кислота, 0,1 н. раствор: 4,946 г As2O3 ч.д.а. растворяют в дистиллированной воде, подкисляют добавлением 0,2 мл концентрированной серной кислоты ч.д.а. и разбавляют дистиллированной водой до 500 мл.
Серная кислота ч.д.а., концентрированная.
Сульфат церия и аммония, 0,02 н. раствор: 13,38 г Ce(NH4)4(SO4)4·4H2O растворяют в дистиллированной воде, подкисленной 44 мл концентрированной серной кислоты ч.д.а., и разбавляют водой до 1 л.
Сульфат церия и аммония, 0,005 М раствор в 3 н. серной кислоте: 3,342 г Ce(NH4)4(SO4)4·4H2O обрабатывают 500 мл дистиллированной воды, прибавляют 80 мл концентрированной серной кислоты ч.д.а. и после полного растворения дополняют дистиллированной водой до 1 л.
Соль Мора, 1,5%-ный раствор: 1,5 г Fe(NH4)2(SO4)2·6H2O растворяют в дистиллированной воде, прибавляют 0,6 мл концентрированной серной кислоты ч.д.а. и разбавляют водой до 100 мл. Применяют всегда свежеприготовленный раствор.
Роданид калия, 4%-ный раствор: 4,0 г KCNS ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Хлорид натрия + мышьяковистая кислота, раствор в 4,5 н. серной кислоте: 58,45 г NaCl ч.д.а. растворяют в 200 мл дистиллированной воды. Отдельно растворяют 4,9460 г As2O3 ч.д.а. в 20 мл 7%-ного раствора едкого кали. Также отдельно смешивают 120 мл концентрированной серной кислоты ч.д.а. и 380 мл дистиллированной воды. Все три раствора соединяют и дополняют водой до 1 л.
Йодид калия, стандартный раствор:
а) запасной: 0,2616 г KJ ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л. Хранят в склянке из коричневого стекла; 1 мл раствора содержит 0,200 мг J-;
б) рабочий I: 50,0 мл запасного раствора разбавляют до 1 л дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,010 мг J-;
в) рабочий II: 10 мл рабочего раствора I разбавляют до 1 л дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,00010 мг J-.
Ход определения
33.6. Вариант А. В реакционную пробирку наливают 10 мл пробы воды, если надо, предварительно разбавленной или сконцентрированной выпариванием, чтобы содержание йодидов в этом объеме было в пределах 0,2 - 0,6 мкг. Одновременно обрабатывают холостую пробу - дистиллированную воду. Прибавляют последовательно 1 мл раствора хлорида натрия, 0,50 мл раствора мышьяковистой кислоты и 0,50 мл концентрированной серной кислоты. Смесь перемешивают и пробирки погружают в водяную баню с температурой 30 +/- 0,5 °C. Одновременно в эту же баню погружают пробирку с раствором сульфата церия и аммония. Когда установится температура, отмеряют 1 мл раствора сульфата церия и аммония, прибавляют его к исследуемой смеси и немедленно перемешивают. Начиная с этого момента, отсчитывают секундомером время. По истечении 15 +/- 0,1 мин пробирку вынимают из бани и немедленно прибавляют 1 мл раствора соли Мора. После перемешивания и исчезновения желтой окраски ионов четырехвалентного церия приливают 1 мл раствора роданида. Смесь снова перемешивают, помещают в баню и через 30 мин от момента прибавления раствора роданида измеряют оптические плотности пробы и холостого раствора и по калибровочной кривой находят содержание йодид-ионов.
Калибровочная кривая
33.7. В ряд мерных колб емкостью 100 мл отмеряют 0 - 20,0 - 40,0 - 60,0 - 80,0 мл стандартного рабочего раствора II и доливают до меток дистиллированной водой. Растворы соответствуют концентрациям 0 - 0,20 - 0,040 - 0,060 и 0,080 мг/л йодид-ионов. Хорошо перемешивают, отмеривают по 10 мл приготовленных стандартных растворов, переносят в набор реакционных пробирок и дальше продолжают, как указано в ходе определения. Значения измеренных величин оптической плотности наносят на график против соответствующих концентраций йодид-ионов. Зависимость нелинейна. Калибровочную кривую надо строить всегда заново после приготовления нового раствора мышьяковистой кислоты или сульфата церия и аммония. Рекомендуется калибровочную кривую часто проверять.
Вариант Б. Пробу воды обрабатывают по варианту А. По истечении требуемого времени реакции пробирку погружают в ледяную баню, где ее оставляют на 10 мин. После этого немедленно измеряют оптическую плотность. Подобным же способом проводят холостой опыт с дистиллированной водой и по калибровочной кривой находят содержание йодид-ионов.
Калибровочная кривая
33.8. Стандартные растворы готовят по варианту А. После этого их обрабатывают по варианту Б. Значения оптической плотности наносят на график против соответствующих концентраций йодид-ионов. Зависимость нелинейная. Калибровочную кривую рекомендуется часто проверять.
Расчет. Содержание йодидов (x) в мг/л вычисляют по формуле

где c - концентрация йодид-ионов, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой на определение, мл.
Округление результатов
Диапазон, мг/л | 0,0 - 0,010 | 0,010 - 0,100 | 0,10 - 1,00 |
Округление, мг/л | 0,0001 | 0,001 | 0,01 |
Йодометрическое определение
33.9. Йодометрическим способом йодиды определяют после окисления их до йодатов. Окисление проводят бромной водой; избыточный бром удаляют добавлением муравьиной кислоты.
При анализе 1 л пробы можно определить этим способом содержание йодидов, начиная от 0,02 мг/л. Пробы с более низким содержанием йодидов предварительно концентрируют упариванием.
Мешающие влияния
33.10. Определению мешает присутствие органических веществ. Их удаление предусмотрено ходом анализа.
Аппаратура
33.11. Платиновые чашки.
Водяная баня.
Реактивы
33.12. Карбонат натрия, твердый ч.д.а.
Этиловый спирт ч.д.а., 96%-ный.
Буферный раствор, pH = 6,8 (однозамещенный фосфат натрия + двузамещенный фосфат натрия + пирофосфат натрия): 20 г NaH2PO4 ч.д.а., 20 г Na2HPO4 ч.д.а. и 20 г Na4P2O7 ч.д.а., высушенных при 110 °C, растворяют в дистиллированной воде и разбавляют до 250 мл.
Бром ч.д.а., насыщенный водный раствор.
Муравьиная кислота, примерно 2 н. раствор: 92 мл HCOOH ч.д.а. разбавляют дистиллированной водой до 1 л.
Йодид калия, 1 н. раствор: 16,6 г KJ ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Тиосульфат натрия, 0,01 н. раствор, приготовление см. стр. 186.
Крахмал, 0,5%-ный раствор, приготовление см. стр. 63.
Ход определения
33.13. Для определения отбирают такой объем пробы, чтобы содержание в нем йодидов было в пределах 0,02 - 2 мг. Пробу подщелачивают твердым карбонатом натрия и постепенно выпаривают в платиновой чашке на водяной бане до малого объема. Прибавляют 1 мл 10%-ного раствора сульфата цинка, перемешивают и затем выпаривают досуха. Остаток после выпаривания прокаливают при температуре 600 °C. По охлаждении остаток в чашке несколько раз экстрагируют этиловым спиртом. Растворение ускоряют перемешиванием взвеси стеклянной палочкой. Соединенные спиртовые экстракты количественно фильтруют через маленький фильтр в другую платиновую чашку и фильтр промывают этиловым спиртом. Спиртовой экстракт выпаривают досуха на водяной бане и остаток количественно смывают с помощью 30 мл дистиллированной воды в колбу для титрования, снабженную притертой пробкой. Прибавляют 1 мл буферного раствора и 5 мл бромной воды. Перемешивают и сейчас же прибавляют 2 мл 2 н. раствора муравьиной кислоты. Смесь перемешивают в течение 2 мин, прибавляют 1 мл 1 н. раствора йодида калия, снова перемешивают и через 1 мин титруют выделенный йод из микробюретки 0,01 н. раствором тиосульфата с индикатором крахмалом. На появляющуюся через некоторое время фиолетовую окраску не обращают внимания. Одновременно проводят холостой опыт с 30 мл дистиллированной воды и всеми реактивами.
Расчет. Содержание йодид-ионов (x) в мг/л вычисляют по формуле

где a - объем 0,01 н. раствора тиосульфата, израсходованного при титровании пробы, мл;
b - объем 0,01 н. раствора тиосульфата, израсходованного при холостом опыте, мл;
V - объем пробы, взятой на определение, мл;
k - поправка для проведения концентрации раствора тиосульфата к точно 0,01 н.;
21,15 - эквивалент йода в этой реакции.
Округление результатов
Диапазон, мг/л | 0,002 <*> - 0,100 | 0,100 - 0,200 | 0,20 - 0,30 | 0,50 - 1,00 |
Округление, мг/л | 0,002 | 0,005 | 0,01 | 0,02 |
--------------------------------
Общие положения
34.1. Мышьяк обычно находится в воде в виде арсенатов. В подземных водах арсенаты присутствуют редко. Мышьяк входит в состав некоторых минеральных вод, а также шахтных вод. В поверхностные воды мышьяк попадает из сточных вод обогатительных фабрик, из отходов производства красителей, кожевенных заводов, основной химической промышленности и металлургических заводов. Мышьяк может содержаться в смывах с площадей земли, где применяли инсектициды, содержащие мышьяк.
Для определения мышьяка в питьевой, поверхностной и сточной водах приводится колориметрический метод с диэтилдитиокарбаминатом серебра. При обработке 50 мл пробы можно определить даже 0,05 мг As в 1 л воды. Для определения еще меньших количеств мышьяка пробу концентрируют выпариванием или соосаждением мышьяка с гидроокисью железа.
Пробы консервируются прибавлением 5 мл HCl на 1 л воды.
Результаты определения приводятся в миллиграммах мышьяка на 1 л воды.
Качественное определение
34.2. В пробирку с анализируемой водой прибавляют несколько капель концентрированной серной кислоты и на кончике ножа вводят немного порошкообразного цинка. Пробирку закрывают ватной пробкой, смоченной в растворе нитрата свинца, и на вату кладут кристаллик нитрата серебра, смоченного водой. В присутствии мышьяка нитрат серебра сначала окрашивается в желтый цвет, потом чернеет. Определению мешает сероводород. Положительная реакция получается при концентрации мышьяка, превышающей 0,1 мг/л.
Колориметрическое определение
с диэтилдитиокарбаминатом серебра
34.3. Мышьяковистый водород (арсин), образовавшийся при действии на соединения мышьяка водорода в момент выделения, реагирует с диэтилдитиокарбаминатом серебра в присутствии пиридина. Раствор окрашивается в красный цвет, интенсивность которого зависит от количества мышьяка.
Мешающие влияния
34.4. Определению мешает сероводород, который также вступает в реакцию с указанным реактивом и дает подобную же окраску. Мешающее влияние сероводорода устраняют помещением на пути проходящих газов фильтрационной трубки с бумагой, пропитанной ацетатом свинца, или минерализацией пробы, проводимой для удаления органических веществ (см. ниже).
Сурьмянистый водород в концентрациях, превышающих 0,1 мг/л, дает розовую окраску. В обычных анализах с ним можно не считаться.
Определению мешает высокое содержание органических веществ. Их удаляют минерализацией. Пробу воды, содержащую от 0,002 до 0,04 мг мышьяка, выпаривают с 2 - 3 мл концентрированной серной кислоты, 5 мл концентрированной азотной кислоты и 0,5 мл 30%-ной перекиси водорода до появления белого дыма серной кислоты. После охлаждения прибавляют 5 мл дистиллированной воды, содержимое чашки переносят в колбу прибора и доводят дистиллированной водой приблизительно до 50 мл. Такую обработку можно применить и для концентрирования мышьяка в пробе.
При анализе проб с низким содержанием органических веществ концентрирование мышьяка достигается соосаждением его с гидроокисью железа. Необходимый объем пробы (100 - 1000 мл), содержащий не менее 0,002 мг мышьяка, помещают в делительную воронку с широким отверстием втулки крана, прибавляют 0,5 - 1 мл раствора хлорида железа (145 г безводного FeCl3 ч.д.а. в 1 л дистиллированной воды) и осторожно нейтрализуют разбавленным (1:4) раствором аммиака до появления осадка. Жидкость в воронке слегка перемешивают в течение 15 мин, дают осадку собраться на дне воронки, после чего, открыв кран делительной воронки, быстро удаляют из нее весь осадок, содержащий мышьяк. Осаждение повторяют таким же способом. Полученный осадок растворяют в необходимом количестве разбавленной (1:4) соляной кислоты. Если не весь осадок растворится, раствор фильтруют, собирая фильтрат в банку прибора, и разбавляют его дистиллированной водой примерно до 50 мл.
Аппаратура
34.5. Стеклянный прибор, состоящий из следующих частей: конической колбы емкостью 100 - 150 мл с притертым горлом, притертого к этой колбе удлинителя с боковым отводом и с воронкой, снабженной краном, достигающей до дна колбы; стеклянной фильтрационной трубки длиной примерно 5 см; вводной трубки в виде буквы L, суженной на одном конце; поглотительного сосуда с внутренним диаметром 1 см, высотой 10 см, со стеклянной спиральной вставкой. Притертые части прибора скрепляются пружинами.
Фотометр, светофильтр  .
.
.Реактивы
34.6. Диэтилдитиокарбаминат серебра, поглотительный раствор в пиридине: 2,25 г диэтилдитиокарбамината натрия (купраля) растворяют в 100 мл дистиллированной воды и по частям осаждают раствором нитрата серебра, приготовленным растворением 1,7 г AgNO3 ч.д.а. в 100 мл дистиллированной воды. Осадок, который должен быть светло-желтым, обрабатывают дистиллированной водой, фильтруют и высушивают в эксикаторе при пониженном давлении. В 200 мл свежеперегнанного пиридина растворяют 1 г приготовленной соли.
Йодид калия, 15%-ный раствор.
Соляная кислота ч.д.а. без мышьяка, концентрированная.
Хлорид олово (II), 40%-ный раствор в концентрированной соляной кислоте ч.д.а. (без мышьяка).
Цинк ч.д.а. (без мышьяка), гранулированный или порошкообразный.
Бумажки, пропитанные раствором ацетата свинца. Полоски фильтровальной бумаги пропитывают 10%-ным раствором ацетата свинца и высушивают.
Арсенит натрия, стандартный раствор:
а) запасной: 0,1320 г As2O3 ч.д.а. растворяют в 10 мл 1 н. раствора едкого натра, прибавляют 10 мл 1 н. раствора серной кислоты и доводят дистиллированной водой до 1 л; 1 мл раствора содержит 0,100 мг As;
б) рабочий: 10,0 мл запасного раствора разбавляют дистиллированной водой до 1 л. Применяют всегда свежеприготовленный раствор; 1 мл раствора содержит 0,001 мг As.
Калибровочная кривая
34.7. Приготовляют шкалу стандартов, содержащих 0 - 2,0 - 5,0 - 10,0 - 20,0 - 30,0 - 40,0 мл рабочего стандартного раствора арсенита, и их объемы доводят дистиллированной водой до 50 мл. Полученные растворы соответствуют концентрациям 0 - 0,040 - 0,100 до 0,80 мг/л мышьяка. Их обрабатывают описанным ниже способом. Из полученных значений оптической плотности вычитают оптическую плотность холостого определения и наносят их на график против соответствующих концентраций мышьяка.
Ход определения
34.8. В начале работы проверяют применяемые реактивы на содержание в них мышьяка, проводя холостое определение с дистиллированной водой.
В колбу прибора наливают 50 мл первоначальной пробы или предварительно сконцентрированной так, чтобы она содержала в этом объеме от 0,002 до 0,040 мг As, и подкисляют добавлением 15 мл соляной кислоты. Можно подкислять и серной кислотой, прибавляя 20 мл разбавленной (1:7) H2SO4. Прибавляют 6 мл раствора йодида калия и 0,5 мл раствора хлорида олова (II). Смесь перемешивают и дают постоять 15 мин. В фильтрационную трубку помещают сухую бумажку, пропитанную ацетатом свинца, а в поглотительный сосуд наливают 15 мл поглощающего раствора. Вводную трубку соединяют с фильтрационной и с удлинителем. К пробе прибавляют 5 г гранулированного или 2 - 3 г порошкообразного цинка, сейчас же присоединяют удлинитель и конец вводной трубки погружают в поглотительный раствор. Притертые соединения скрепляют пружинами. Водород образуется в течение 60 мин. В случае замедленного образования водорода усиливают его выделение прибавлением 0,5 мл раствора хлорида олова и 5 - 15 мл концентрированной HCl. После прекращения процесса образования водорода поглотительный раствор наливают в колориметрическую кювету и определяют оптическую плотность. Вычитают значение ее для холостого определения с дистиллированной водой и по калибровочной кривой находят содержание мышьяка.
Расчет. Содержание мышьяка (x) в мг/л вычисляют по формуле

где c - концентрация мышьяка, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл;
50 - объем, до которого разбавлена проба, мл.
Округление результатов
Диапазон, мг/л | 0,005 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,00 |
Округление, мг/л | 0,005 | 0,01 | 0,02 | 0,05 |
Общие положения
35.1. Нитраты встречаются почти во всех видах вод. В поверхностных и подземных водах количество их обычно незначительно, однако в некоторых типах подземных вод концентрация нитратов может быть высока. При концентрациях нитратов 0,5 - 50 мг/л в питьевых и поверхностных водах наиболее удобным является колориметрический метод определения с фенолдисульфоновой кислотой. Хорошие результаты дает и колориметрический метод с салицилатом натрия при концентрациях 0,1 - 20 мг/л.
Большие количества нитратов можно определять указанными методами при соответствующем разбавлении пробы.
Для сведения приведен также колориметрический метод, который состоит в восстановлении нитратов до нитритов гидразином; метод применим для концентрации  0,01 - 2,0 мг/л.
0,01 - 2,0 мг/л.
Если проба не была обработана в день отбора, ее сохраняют в холодильнике или консервируют добавлением 1 мл концентрированной серной кислоты на 1 л пробы или 2 - 4 мл хлороформа.
Результаты определений выражаются в мг-экв/л или в мг/л: 1 мг-экв 
 ; 1 мг
; 1 мг 
 .
.
Колориметрическое определение нитратов
с фенолдисульфоновой кислотой
35.2. Определение основано на образовании желтого окрашивания при реакция нитратов с фенолдисульфоновой кислотой. Без разбавления этим методом можно определять от 0,5 до 50 мг  в 1 л воды.
в 1 л воды.
При концентрации  в пределах 2 - 50 мг/л и отсутствии мешающих влияний точность определения +/- 0,5 мг/л.
в пределах 2 - 50 мг/л и отсутствии мешающих влияний точность определения +/- 0,5 мг/л.
Мешающие влияния
35.3. Определению мешают хлориды. Концентрация их в пробе не должна превышать 10 мг/л. Это достигается соответствующим разбавлением. Мешающее влияние хлоридов можно также устранить добавлением сульфата серебра (см. ниже).
Однако при анализе некоторых вод избыток ионов серебра вызывает окрашивание или образование мути. Для подщелачивания пробы применяют едкое кали или аммиак.
Если для осаждения хлоридов был применен сульфат серебра, то подщелачивать в дальнейшем раствор едким кали нельзя, так как этот реактив вызывает коричневое окрашивание. Тогда вместо него следует применять раствор аммиака.
При содержании нитритов более 0,7 мг/л  получаются повышенные результаты. Окрашенные соединения не должны присутствовать.
получаются повышенные результаты. Окрашенные соединения не должны присутствовать.
Удаление хлоридов. Определяют в пробе содержание хлоридов. К 100 мл пробы прибавляют эквивалентное количество раствора сульфата серебра (4,40 г Ag2SO4 ч.д.а., свободного от нитритов, растворяют в дистиллированной воде и доводят до 1 л; 1 мл раствора соответствует 1 мг Cl-). Осадок хлорида серебра отфильтровывают или центрифугируют после предварительного нагревания смеси. Целесообразно оставить смесь стоять на ночь в темном месте.
Удаление окраски: к 150 мл пробы прибавляют 3 мл суспензии гидроокиси алюминия (125 г сульфата алюминия и калия или сульфата алюминия и аммония растворяют в 1 л дистиллированной воды, нагревают до 60 °C и постепенно прибавляют 55 мл концентрированного раствора аммиака при постоянном перемешивании; после часового отстаивания осадок переносят в большой стакан и промывают декантацией дистиллированной водой до исчезновения реакции на свободный аммиак, хлориды, нитриты и нитраты), тщательно перемешивают, оставляют несколько минут стоять и затем фильтруют.
Окисление нитритов: к 100 мл пробы прибавляют 1 мл 1 н. раствора H2SO4 и перемешивают. Затем по каплям прибавляют при перемешивании необходимое количество 0,1 н. раствора перманганата калия или 3%-ного раствора перекиси водорода. Перманганат добавляется в таком количестве, чтобы розовая окраска не исчезла в течение 15 мин. Из результатов определения вычитают содержание нитритов. Они определяются в отдельной пробе.
Аппаратура
35.4. Фотометр, светофильтр  . Для повышения верхнего предела определяемой концентрации можно производить измерения и при других длинах волн, например при 480 нм.
. Для повышения верхнего предела определяемой концентрации можно производить измерения и при других длинах волн, например при 480 нм.
. Для повышения верхнего предела определяемой концентрации можно производить измерения и при других длинах волн, например при 480 нм.Кюветы длиной 1 - 5 см или набор цилиндров Несслера емкостью 50 или 100 мл.
Реактивы
35.5. Фенолдисульфоновая кислота, раствор в серной кислоте: 25 г фенола ч.д.а. (препарат не должен быть окрашен) растворяют в 150 мл концентрированной серной кислоты. Прибавляют 75 мл дымящей серной кислоты (олеум с 15% SO3), тщательно перемешивают и нагревают с обратным холодильником в течение 2 ч на кипящей водяной бане.
Аммиак ч.д.а., концентрированный раствор.
Едкое кали, приблизительно 12 н. раствор: 673 г KOH ч.д.а. растворяют в дистиллированной воде и доводят до 1 л.
Комплексон III, аммиачный раствор: 50 г комплексона III растирают с 20 мл дистиллированной воды до получения пасты, которую растворяют в 60 мл концентрированного раствора аммиака.
Нитрат калия, стандартный раствор: 0,1631 г KNO3 ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде, прибавляют 1 мл хлороформа и доводят водой до 1 л; 1 мл содержит 0,100 мг  .
.
Калибровочная кривая
35.6. На водяной бане выпаривают досуха 50 мл стандартного раствора; сухой осадок растворяют в 2 мл кислого раствора фенолдисульфоновой кислоты и доводят дистиллированной водой до 100 мл.
Для визуального колориметрирования, производимого в цилиндрах Несслера, в ряд цилиндров емкостью по 50 мл отмеривают, например, 0,1 - 0,3 - 0,5 - 0,7 - 1,0 - 2,0 - 5,0 - 10 - 15 - 20 и 30 мл окрашенного стандартного раствора (что отвечает 0 - 0,1 - 0,3 до 30 мг  ), прибавляют 2 мл раствора фенолдисульфоновой кислоты и такое же количество раствора NH4OH или KOH, какое прибавлялось к пробе.
), прибавляют 2 мл раствора фенолдисульфоновой кислоты и такое же количество раствора NH4OH или KOH, какое прибавлялось к пробе.
Объем раствора в цилиндрах доводят дистиллированной водой до метки и перемешивают. Приготовленные таким образом окрашенные стандартные растворы для сравнения могут сохраняться несколько недель без изменения окраски.
Калибровочную кривую для колориметрического определения получают, измеряя оптическую плотность окрашенных стандартных растворов, приготовленных, как указано выше, при описании визуального колориметрирования. Из найденных значений вычитают оптическую плотность холостой пробы. Найденные значения оптической плотности откладывают на графике против соответствующих концентраций нитратов.
Ход определения
35.7. Прозрачную пробу объемом 100 мл или меньше с содержанием не более 5 мг  нейтрализуют до pH = 7, переливают в фарфоровую чашку и выпаривают на кипящей водяной бане досуха. К сухому остатку прибавляют 2 мл раствора фенолдисульфоновой кислоты и размешивают стеклянной палочкой до полного растворения. Если потребуется, смесь слегка нагревают на водяной бане. Прибавляют 20 мл дистиллированной воды и приливают при помешивании 6 - 7 мл концентрированного раствора аммиака или 5 - 6 мл 12 н. раствора едкого кали. Если при этом выделяются гидроокиси присутствующих металлов, их удаляют фильтрованием через фильтрующий тигель или же прибавляют по каплям аммиачный раствор комплексона III до полного растворения осадка. Фильтрат или прозрачный раствор переносят в мерную колбу емкостью 50 или 100 мл, доводят дистиллированной водой до метки и содержимое колбы перемешивают. Окрашенный раствор колориметрируют. Из найденного значения вычитают оптическую плотность холостого опыта, проведенного с дистиллированной водой, так же как и анализ пробы, и по калибровочной кривой находят содержание нитрат-ионов.
нейтрализуют до pH = 7, переливают в фарфоровую чашку и выпаривают на кипящей водяной бане досуха. К сухому остатку прибавляют 2 мл раствора фенолдисульфоновой кислоты и размешивают стеклянной палочкой до полного растворения. Если потребуется, смесь слегка нагревают на водяной бане. Прибавляют 20 мл дистиллированной воды и приливают при помешивании 6 - 7 мл концентрированного раствора аммиака или 5 - 6 мл 12 н. раствора едкого кали. Если при этом выделяются гидроокиси присутствующих металлов, их удаляют фильтрованием через фильтрующий тигель или же прибавляют по каплям аммиачный раствор комплексона III до полного растворения осадка. Фильтрат или прозрачный раствор переносят в мерную колбу емкостью 50 или 100 мл, доводят дистиллированной водой до метки и содержимое колбы перемешивают. Окрашенный раствор колориметрируют. Из найденного значения вычитают оптическую плотность холостого опыта, проведенного с дистиллированной водой, так же как и анализ пробы, и по калибровочной кривой находят содержание нитрат-ионов.
Расчет. Содержание нитрат-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
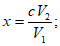

где c - концентрация нитрат-ионов, найденная по калибровочной кривой или сравнением со стандартными растворами, мг/л;
V1 - объем пробы, взятой для анализа, мл;
V2 - объем окрашенной пробы мл (50 или 100);
62,0 - эквивалент  .
.
Округление результатов
Диапазон, мг/л | 0,10 - 1,00 | 1,0 - 2,0 | 2,0 - 5,0 | 5,0 - 10,0 |
Округление: | ||||
мг/л | 0,05 | 0.1 | 0,2 | 0,5 |
мг-экв/л | 0,001 | 0,002 | 0,005 | 0,01 |
Колориметрическое определение нитратов с салицилатом натрия
35.8. Определение основано на реакции нитратов с салицилатом натрия в среде серной кислоты, в результате которой образуются окрашенные в желтый цвет соли нитросалициловой кислоты. Без разбавления можно определять от 0,1 до 20 мг/л  .
.
Мешающие влияния
35.9. Коллоидные органические и окрашенные вещества, присутствующие в пробе, мешают определению. Их удаляют так же, как и при определении нитратов с фенолдисульфоновой кислотой (см. стр. 177).
Если сухой остаток после прибавления серной кислоты окрасится присутствующими растворенными органическими веществами, то этот метод неприменим.
Определению мешают хлориды в количествах, превышающих 200 мг/л. Их удаляют способом, изложенным при определении нитратов с фенолдисульфоновой кислотой (см. стр. 176).
Железо мешает определению в количестве более 5 мг/л. Мешающие влияния большой концентрации железа или других катионов можно устранить фильтрованием через катионит.
Нитриты влияют на определение при содержании их, превышающем 1 - 2 мг/л. Содержание 20 мг 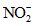 1 л повышает найденный результат при определении нитратов на 1 мг/л. Нитрит-ионы в количестве более 2 мг/л следует удалять выпариванием 20 мл пробы досуха на водяной бане с добавлением 0,05 г сульфата аммония.
1 л повышает найденный результат при определении нитратов на 1 мг/л. Нитрит-ионы в количестве более 2 мг/л следует удалять выпариванием 20 мл пробы досуха на водяной бане с добавлением 0,05 г сульфата аммония.
Аппаратура
ИС МЕГАНОРМ: примечание. Нумерация пунктов дана в соответствии с официальным текстом документа. |
35.9. Фотометр, светофильтр  .
.
.Кюветы толщиной 1 - 5 см или набор цилиндров Несслера емкостью 50 мл.
Водяная баня.
Реактивы
35.10. Салицилат натрия, 0,5%-ный водный раствор (всегда свежеприготовленный).
Серная кислота ч.д.а., концентрированная, свободная от нитратов.
Едкий натр, приблизительно 10 н. раствор: 400 г NaOH ч.д.а. растворяют в дистиллированной воде и после охлаждения доводят до 1 л.
Нитрат калия, стандартный раствор:
а) запасной (приготовление см. раздел 35.2); 1 мл раствора содержит 0,100 мг  ;
;
б) рабочий: разбавляют 10,0 мл запасного раствора дистиллированной водой до 100 мл; всегда применяют свежеприготовленный раствор; 1 мл раствора содержит 0,010 мг  .
.
Калибровочная кривая
35.11. Калибровочную кривую строят обработкой ряда стандартных растворов, содержащих 0 - 0,5 - 1,0 - 2,0 - 5,0 - 10,0 - 20 мг  в 1 л. Для этого отмеривают 0 - 0,5 - 1,0 и т.д. мл рабочего раствора нитрата калия, доводят объем дистиллированной водой до 10 мл и заканчивают, как описано ниже.
в 1 л. Для этого отмеривают 0 - 0,5 - 1,0 и т.д. мл рабочего раствора нитрата калия, доводят объем дистиллированной водой до 10 мл и заканчивают, как описано ниже.
Для визуальной колориметрии в цилиндрах Несслера приготавливают окрашенные стандартные растворы объемом в 50 мл, обработанные, как указано ниже, например, для ряда 0 - 0,2 - 0,5 - 1,0 - 2,0 - 4,0 - 6,0 - 8,0 - 10,0 мг  в 1 л (0 - 0,2 - 0,5 и т.д. мл рабочего раствора доводят до 10 мл дистиллированной водой).
в 1 л (0 - 0,2 - 0,5 и т.д. мл рабочего раствора доводят до 10 мл дистиллированной водой).
Ход определения
35.12. К 10 мл пробы прибавляют 1 мл раствора салицилата натрия и выпаривают в фарфоровой чашке на водяной бане досуха. После охлаждения сухой остаток увлажняют 1 мл серной кислоты и оставляют стоять 10 мин. Содержимое чашки разбавляют дистиллированной водой, переносят количественно в мерную колбу емкостью 50 мл, прибавляют 7 мл 10 н. раствора едкого натра, доводят дистиллированной водой до метки и тщательно перемешивают. После охлаждения до комнатной температуры вновь доводят до метки и окрашенный раствор колориметрируют. В течение 10 мин после прибавления раствора едкого натра окраска не изменяется. Из найденных значений оптической плотности вычитают оптическую плотность холостой пробы (приготовленной тем же способом) с дистиллированной водой.
Расчет. Расчет такой же, как и при определении нитратов с фенолдисульфоновой кислотой.
Округление результатов. Результаты округляют так же, как и при определении нитратов с фенолдисульфоновой кислотой.
Определение нитратов после восстановления до аммиака
35.13. Определение основано на восстановлении нитратов до аммиака водородом в момент выделения, образующимся при реакции едкого кали со сплавом Деварда. Аммиак отгоняют из реакционной смеси и улавливают в приемнике с серной кислотой, в котором его затем определяют либо объемным методом, либо колориметрическим. Колориметрический метод следует применять при концентрации нитратов ниже 10 мг/л; при концентрации выше 5 мг/л  определение производят объемным методом.
определение производят объемным методом.
Мешающие влияния
35.14. Результаты определения искажаются присутствием аммиака и нитритов. Аммиак или образующие его в условиях определения вещества устраняются выпариванием щелочной пробы, как описано в ходе определения. Нитриты необходимо определить отдельно, и полученный результат вычесть из результата определения нитратов.
Аппаратура
35.15. Перегонный аппарат (перегонная колба круглодонная емкостью 500 - 1000 мл, насадка с делительной воронкой и каплеуловителем, холодильник шариковый длиной 300 мм с длинным концом).
Аппаратура для определения аммиака (см. стр. 103).
Реактивы
35.16. Едкий натр, 25%-ный раствор: 250 г NaOH ч.д.а. растворяют в 1250 мл дистиллированной воды, прибавляют около 0,5 г сплава Деварда и упариванием доводят объем до 1 л.
Сплав Деварда (порошок).
Дважды дистиллированная вода, свободная от аммиака.
Реактив Несслера (приготовление см. стр. 100).
Серная кислота, 0,02 н. раствор (приготовление см. стр. 103).
Едкий натр, 0,02 н. раствор (приготовление см. стр. 103).
Смешанный индикатор (метиловый красный + метиленовая синяя) (приготовление см. стр. 103).
Ход определения
35.17. К 200 мл пробы, налитой в перегонную колбу, прибавляют 20 мл 25%-ного раствора едкого натра и объем смеси упаривают наполовину. После охлаждения прибавляют 200 мл дважды дистиллированной воды и около 0,5 г сплава Деварда. Колбу быстро соединяют изогнутой насадкой с холодильником. Конец холодильника должен быть погружен в жидкость, находящуюся в приемнике. Смесь в перегонной колбе нагревают сначала слабо и только после окончания выделения водорода доводят ее до кипения. После отгонки около 150 мл жидкости перегонку прекращают. Содержимое приемника разбавляют дважды дистиллированной водой до 200 мл.
Обработку и определение аммиака в дистилляте (колориметрически или титрованием) производят так же, как и при определении аммиака (стр. 99). Одновременно с анализом пробы определяют количество титрованного раствора, израсходованного на холостой опыт с дважды дистиллированной водой.
Расчет. Содержание нитратов в мг/л (x) или в мг-экв/л (y) рассчитывают по формулам:
x = 3,44a - 1,35b;
y = 0,01613(3,44a - 1,35b),
где a - концентрация аммиака, выраженная в мг/л 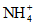 ;
;
b - концентрация нитритов  , найденная в отдельной пробе, мг/л.
, найденная в отдельной пробе, мг/л.
Содержание нитратов, полученных при определении объемным методом, рассчитывается по формулам:


где a1 - количество 0,02 н. раствора едкого натра, израсходованного на холостой опыт при титровании 25,0 мл 0,02 н. раствора серной кислоты, мл;
a2 - количество 0,02 н. раствора едкого натра, израсходованного на взятую пробу, мл;
V - объем пробы, взятой на определение, мл;
k - поправка для проведения нормальности раствора едкого натра к точно 0,02 н.;
b - см. выше.
Округление результатов
Диапазон, мг/л | 2,0 - 5,0 | 5,0 - 10,0 | 10 - 20 | 20 - 50 |
Округление: | ||||
мг | 0,2 | 0,5 | 1 | 2 |
мг-экв | 0,05 | 0,01 | 0,02 | 0,05 |
Общие положения
36.1. Нитриты являются промежуточным продуктом микробиального окисления аммиака или восстановления нитратов. Их присутствие свидетельствует о фекальном загрязнении воды. В поверхностных водах нитриты быстро переходят в нитраты. Они присутствуют в концентрациях от нескольких микрограммов до десятых долей миллиграмма в 1 л. В большем количестве они находятся в некоторых промышленных и биологически очищенных сточных водах.
Для определения нитритов в питьевых и поверхностных водах приведен колориметрический метод с сульфаниловой кислотой и альфа-нафтиламином.
Ввиду нестойкости нитритов необходимо их определять тотчас же после отбора пробы. Если это невозможно, пробу консервируют добавлением 1 мл концентрированной H2SO4 или 2 - 4 мл хлороформа на 1 л. Можно также охлаждать пробу до 3 - 4 °C.
Результаты, полученные при определении, выражаются в миллиграммах нитрит-ионов на 1 л, а при больших концентрациях - в мг-экв на 1 л; 1 мг/л NO2 = 0,02174 мг-экв/л NO2; 1 мг-экв/л NO2 = 40,005 мг/л NO2.
Качественное определение
36.2. К 10 мл пробы прибавляют 1 мл раствора сульфаниловой кислоты и 1 мл раствора альфа-нафтиламина (приготовление см. стр. 184). В присутствии нитритов появляется розовая или красно-фиолетовая окраска. Чувствительность около 0,01 мг нитритов в 1 л воды.
Колориметрическое определение с сульфаниловой кислотой
и альфа-нафтиламином
36.3. Определение основано на диазотировании сульфаниловой кислоты присутствующими в пробе нитритами и реакции полученной соли с альфа-нафтиламином с образованием красно-фиолетового азокрасителя. Интенсивность окраски пропорциональна концентрации нитритов. Протекание реакции в значительной степени зависит от pH среды.
Без разбавления пробы можно определять визуально от 0,002 до 0,025 мг  в 1 л; колориметрически в зависимости от примененного фотометра - от 0,001 до 0,6 мг/л. Точность определения +/- 0,002 мг/л.
в 1 л; колориметрически в зависимости от примененного фотометра - от 0,001 до 0,6 мг/л. Точность определения +/- 0,002 мг/л.
Мешающие влияния
36.4. Определению мешают взвешенные вещества и мутность воды. Поэтому перед анализом пробу необходимо профильтровать. Если мутность фильтрованием не устраняется и сильно загрязненные воды содержат коллоидные вещества, необходимо пробу осветлить коагулированием гидроокисью алюминия: к 100 мл пробы прибавляют около 0,5 г активного угля, 1 мл 12,5%-ного сульфата алюминия и калия, раствор аммиака до получения pH = 5,8. После взбалтывания дают осадку осесть до полного осветления пробы. Фильтруют через сухой плотный фильтр ("синяя лента"). Для определения берут часть фильтрата. Осветление можно производить также взбалтыванием 100 мл пробы с 2 мл суспензии гидроокиси алюминия. (Приготовление суспензии см. стр. 177).
В анализируемой пробе не должны присутствовать сильные окислители или восстановители, которые мешают определению.
Трехвалентное железо, двухвалентная ртуть, серебро, висмут, трехвалентная сурьма, свинец, трехвалентное золото, хлороплатинаты и метаванадаты мешают определению, так как выпадают в осадок. Влияния их устраняют соответствующим разбавлением.
Двухвалентная медь занижает результаты вследствие каталитического распада диазотированной сульфаниловой кислоты. В этих случаях также пробу разбавляют.
Определению мешает окраска воды. Слабое окрашивание и незначительная мутность питьевых и поверхностных вод учитываются вычитанием оптической плотности холостой пробы, к которой добавляют раствор сульфаниловой кислоты.
Определению мешает и трихлорамин. При прибавлении реактивов в обратном порядке его мешающее влияние можно в некоторой степени снизить.
Аппаратура
36.5. Фотометр, светофильтр  .
.
.Кюветы длиной 1 - 5 см или набор цилиндров Несслера емкостью 50 мл.
Реактивы
36.6. Сульфаниловая кислота, 0,6%-ный раствор: 6,0 г сульфаниловой кислоты ч.д.а. растворяют в 750 мл горячей дистиллированной воды. К полученному раствору прибавляют 250 мл ледяной уксусной кислоты.
Альфа-нафтиламин, 0,6%-ный раствор: 1,2 г альфа-нафтиламина ч.д.а. растворяют в дистиллированной воде, прибавляют 50 мл ледяной уксусной кислоты и раствор доводят дистиллированной водой до 200 мл. При образовании мути раствор фильтруют через хлопчатобумажную ткань, промытую дистиллированной водой. Раствор сохраняется около 2 - 3 месяцев.
Нитрит натрия, стандартный раствор:
а) запасной: растворяют 0,1497 г NaNO3 ч.д.а., высушенного при 105 °C, в дистиллированной воде (лучше всего в стерилизованной) и доводят до 1 л; раствор консервируют добавлением 1 мл хлороформа и сохраняют в холодном месте; раствор устойчив в течение 1 месяца; 1 мл этого раствора содержит 0,100 мг 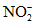 ;
;
б) рабочий I: разбавляют 100 мл запасного раствора дистиллированной водой до 1 л. Раствор должен быть всегда свежеприготовленным; 1 мл этого раствора содержит 0,010 мг  ;
;
в) рабочий II: разбавляют 50 мл рабочего раствора I дистиллированной водой до 1 л. Раствор должен быть всегда свежеприготовленным; 1 мл раствора содержит 0,0005 мг 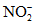 .
.
Калибровочная кривая
36.7. Для построения калибровочной кривой берут серию из шести или восьми стандартных растворов соответственно применяемому прибору с концентрацией нитрит-ионов в пределах от 0 до 0,60 мг/л. Значения оптической плотности наносят на график соответственно концентрациям нитрит-ионов.
При визуальном определении одновременно с пробой готовят серию стандартных растворов в цилиндрах Несслера. В цилиндры емкостью 50 мл отмеривают пипеткой 0 - 0,20 - 0,50 - 1,0 - 1,5 - 2,0 и 2,5 мл стандартного рабочего раствора II и объемы доводят дистиллированной водой до 50 мл. Далее поступают так же, как при анализе пробы (см. ниже). Серия стандартных растворов соответствует концентрациям 0,000 - 0,002 - 0,005 - 0,010 - 0,015 - 0,020 и 0,025 мг  в 1 л.
в 1 л.
Ход определения
36.8. Для определения берут 50 мл или меньшее количество профильтрованной пробы и доводят до 50 мл дистиллированной водой. Прибавляют 1 мл раствора сульфаниловой кислоты и смесь тщательно перемешивают. Если проба мутная, определяют колориметрически ее оптическую плотность, значение которой затем вычитают. После пятиминутного стояния прибавляют 1 мл раствора альфа-нафтиламина и смесь снова перемешивают. Пробу колориметрируют или сравнивают с серией стандартных растворов, приготовленных в цилиндрах Несслера, через 40 мин после прибавления раствора альфа-нафтиламина.
Расчет. Содержание нитрит-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:


где c - концентрация, найденная по калибровочной кривой, или результат сравнения со стандартами, мг/л;
V - объем пробы, взятой для определения, мл;
46 - эквивалент нитрит-иона  .
.
Округление результатов
Диапазон, мг/л | 0,002 - 0,050 | 0,050 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 0,5 - 0 0,05 0,001 |
Округление: | ||||
мг/л | 0,002 | 0,005 | 0,01 | 0,02 |
мг-экв/л | - | - | - | - |
Общие положения
37.1. Озон применяется для обеззараживания воды и для устранения некоторых ее нежелательных ингредиентов или свойств. Для контролирования процесса озонирования необходимо определять содержание озона в обработанной воде.
Для определения озона приведены два метода: относительно более точный йодометрический метод и марганцево-о-толидиновый метод (ОТМ). Метод ОТМ, не будучи ни вполне точным, ни специфичным, пригоден, однако, для обычного контроля на производстве. Определение озона невозможно в присутствии хлора, соединений, выделяющих хлор, и двуокиси хлора.
Пробу не разрешается консервировать. Определение следует производить сразу же после отбора пробы.
Результаты определения выражаются в миллиграммах озона в 1 л воды.
Йодометрическое определение
37.2. Определение основано на окислении озоном йодида до йода, который титруют раствором тиосульфата. Пользуясь 0,01 н. раствором последнего, можно определять озон при содержании его 0,05 мг/л и более.
Мешающие влияния
37.3. Озонированная вода может содержать ионы железа (III) или марганца (IV), нитриты, перекиси и другие вещества, мешающие определению. Поэтому озон вытесняют из пробы воздухом или азотом и поглощают его раствором йодида калия. Устойчивость растворов озона падает с повышением температуры и pH.
Аппаратура
37.4. Сосуды для продувки озона и его поглощения из газа емкостью 1000 и 400 мл (высокие, типа дрекселей со стеклянными фильтрующими пластинками, соединенные стеклянными трубками со шлифами или короткими поливинилхлоридными трубками. Использование резиновых соединений недопустимо).
Реактивы
37.5. Йодид калия, 2,5%-ный раствор: 25 г KJ ч.д.а. растворяют в свежепрокипяченной и охлажденной дистиллированной воде и доводят объем до 1 л. Каждый день приготовляют свежий раствор.
Серная кислота, 10%-ный раствор: 54 мл 96%-ной H2SO4 ч.д.а. осторожно приливают примерно к 750 мл дистиллированной воды и после охлаждения доводят объем до 1 л.
Тиосульфат натрия, 0,1 н. раствор: 24,8 г Na2S2O3·5H2O ч.д.а. растворяют в 900 мл дистиллированной воды, прибавляют 0,2 г Na2CO3 ч.д.а. и после растворения доводят объем до 1 л. Поправку к титру раствора определяют следующим образом: в коническую колбу, снабженную притертой пробкой, наливают примерно 100 мл дистиллированной воды, прибавляют 10 мл 15%-ного раствора йодида калия, 5 мл разбавленной (1:4) серной кислоты и 20 мл 0,1 н. раствора бихромата калия (4,9037 г K2Cr2O7 ч.д.а. в 1 л). После перемешивания раствор оставляют стоять 5 мин в темноте и затем титруют раствором тиосульфата, добавляя 1 - 2 мл раствора крахмала в качестве индикатора. Титр проверяют не реже одного раза в неделю.
Тиосульфат натрия, 0,01 н. раствор: 100 мл 0,1 н. раствора разбавляют свежепрокипяченной и охлажденной дистиллированной водой, добавляют 0,2 г Na2CO3 и доводят объем до 1 л.
Поправку к титру раствора определяют так же, как у 0,1 н. раствора, используя 0,01 н. раствор бихромата калия (0,4904 г K2Cr2O7 в 1 л).
Крахмал, 0,5%-ный раствор (приготовление см. стр. 63).
Йод, 0,01 н. раствор: 1,27 г очищенного возгонкой йода растворяют совместно с 2,5 г KJ в 30 - 40 мл дистиллированной воды и разбавляют водой до 1 л. Поправку к титру раствора определяют непосредственно перед его использованием.
Ход определения
37.6. Из пробы воды объемом до 800 мл, отобранной в сосуд для отдувки газа, вытесняют озон азотом или воздухом, пропуская их через стеклянную фильтрующую пластинку со скоростью 0,2 - 1 л/мин в течение не менее 5 мин. Вытесняемый озон направляется в два последовательно соединенных поглотительных сосуда емкостью по 400 мл. Первый сосуд содержит 200 мл раствора йодида калия, второй - 10 мл 0,01 н. раствора тиосульфата и 40 мл дистиллированной воды. По окончании поглощения озона фильтрующую пластинку ополаскивают и содержимое сосуда с тиосульфатом количественно переносят в сосуд с раствором йодида калия. Прибавляют 25 мл разбавленной серной кислоты, раствор крахмала и титруют 0,01 н. раствором йода до появления синей окраски. Если после смешения содержимого обоих сосудов сразу же возникает с крахмалом синяя окраска, определение повторяют с меньшим количеством пробы.
Параллельно с определением озона проводят холостой опыт с дистиллированной водой с целью обнаружить в реактивах загрязнения, выделяющие йод из йодида калия или, наоборот, восстанавливающие выделенный йод. Для этого к 200 мл раствора йодида калия прибавляют 25 мл разбавленной серной кислоты и крахмал. В случае появления синей окраски титруют 0,01 н. раствором тиосульфата натрия до обесцвечивания и затем титруют 0,01 н. раствором йода до появления синей окраски. Объем израсходованного раствора тиосульфата, за вычетом объема израсходованного раствора йода, прибавляют к объему раствора йода, пошедшему на титрование пробы. Если синяя окраска не появится, то прибавляют 0,01 н. раствор йода до ее появления; потом это количество прибавляют к израсходованному на титрование пробы.
Расчет. Содержание озона (x) в мг/л вычисляют по формуле

где a - расход 0,01 н. раствора йода, мл;
k1 - поправка к титру 0,01 н. раствора тиосульфата;
k2 - поправка к титру 0,01 н. раствора йода;
V - объем пробы, взятой для анализа, мл;
0,01 - нормальность титрующих растворов;
24 - эквивалент озона.
Округление результатов. Результаты округляются до 0,05 мг.
Марганцево-о-толидиновый метод определения
37.7. Определение основано на окислении озоном ионов марганца (II) до марганца (IV), который реагирует с о-толидином в кислой среде. Возникающая окраска имеет такой же оттенок, как и при определении хлора с этим реактивом, и пропорциональна концентрации озона. Чувствительность определения примерно 0,02 мг озона в 1 л.
Мешающие влияния
37.8. Определению мешают присутствие во взятой пробе соединений марганца высших его валентностей. Не мешают нитриты и железо (III). На мешающие влияния можно ввести поправку проведением холостого определения в хорошо проаэрированной пробе.
Аппаратура
37.9. Фотометр, светофильтр  .
.
.Кюветы толщиной 2 - 5 см или набор цилиндров Несслера емкостью 100 мл.
Реактивы
37.10. Сульфат марганца (II), 0,36%-ный раствор: 3,6 г MnSO4·2H2O ч.д.а. (4,3 г MnSO4·4H2O или 3,1 г MnSO4·4H2O) растворяют в дистиллированной воде, содержащей 3 мл концентрированной серной кислоты, и разбавляют до 1 л.
о-Толидин, 0,1%-ный раствор: 1,35 г о-толидингидрохлорида ч.д.а. растворяют в 500 мл дистиллированной воды, полученный раствор при постоянном перемешивании приливают к смеси 350 мл дистиллированной воды и 150 л концентрированной соляной кислоты ч.д.а. Реактив хранят в темной склянке.
Калибровочная кривая
37.11. Для определения хлора служит калибровочная кривая или искусственные стандартные растворы (приготовление см. стр. 230).
Ход определения
37.12. В мерный цилиндр или цилиндр Несслера емкостью 100 мл отмеривают 5,0 мл раствора сульфата марганца, доливают пробой до метки и смесь быстро перемешивают. Прибавляют 5 мл раствора о-толидина и снова быстро перемешивают. Измеряют величину оптической плотности или же сравнивают окраску смеси с искусственными стандартными растворами. Можно пользоваться калибровочной кривой или стандартными растворами, предназначенными для определения хлора (см. стр. 230).
Расчет. Содержание озона (x) в мг/л вычисляют по формуле
x = c·0,69,
где c - содержание озона, найденное по калибровочной кривой или путем сравнения со стандартными растворами, выраженное в мг хлора в 1 л;
0,69 - отношение эквивалентов озона и хлора.
Округление результатов. Результаты округляются до 0,05 мг/л.
Общие положения
38.1. Роданиды не являются естественной составной частью вод. Они имеются в некоторых промышленных сточных водах. В промышленных сточных водах роданиды встречаются почти так же часто, как цианиды, и обычно вместе с ними. Определение роданидов надо производить в день отбора пробы или сразу же после отбора осадить сульфиды (см. "Мешающие влияния"). В концентрациях выше 10 мг/л роданиды рекомендуется определять аргентометрически после отделения нерастворимого роданида меди; для малых концентраций рекомендуется колориметрический метод с бензидином и пиридином.
Пробы не консервируют, их надо анализировать в течение одних суток с момента отбора.
Результаты определения приводятся в миллиграммах роданид-ионов в 1 л воды.
Качественное определение
38.2. К 10 мл пробы прибавляют несколько капель концентрированной соляной кислоты и несколько капель 10%-ного раствора железа (III). По появлению красной окраски судят о присутствии роданидов.
Аргентометрическое определение
38.3. Роданиды осаждаются в виде нерастворимого роданида меди (I). Осадок разлагают щелочью, отделяют фильтрованием окислы меди и в фильтрате титруют роданид-ионы аргентометрически с солью железа (III) в качестве индикатора. Метод пригоден для определения в количествах, превышающих 10 мг/л.
38.4. Мешающие влияния. См. ход определения.
Реактивы
38.5. Карбонат свинца ч.д.а., в порошке.
Фенолфталеин, 0,5%-ный раствор (приготовление см. стр. 36).
Серная кислота ч.д.а., разбавленная (1:2).
Квасцы железоаммонийные ч.д.а., насыщенный раствор.
Пиросульфат калия ч.д.а., 20%-ный раствор; применяют всегда свежеприготовленный.
Сульфат меди, 15%-ный раствор: 15 г CuSO4·5H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Гексацианоферрат калия ч.д.а., 10%-ный раствор.
Карбонат натрия, 4%-ный раствор: 40 г Na2CO3 ч.д.а. растворяют в дистиллированной воде и разбавляют до 1 л.
Азотная кислота ч.д.а., разбавленная (1:1).
Нитрат серебра, 0,05 н. раствор (приготовление см. стр. 233).
Ход определения
38.6. В зависимости от предполагаемого содержания роданидов берут для анализа от 50 до 250 мл профильтрованной пробы сточной воды. При наличии сульфидов добавляют, перемешивая, 0,2 - 0,3 г карбоната свинца. Фильтруют через плотный фильтр (синяя лента). От полученного фильтрата отбирают такую аликвотную часть, чтобы в ней содержалось от 2 до 30 мг роданид-ионов, но не больше 200 мл. Если объем отобранной части меньше 100 мл, разбавляют ее дистиллированной водой до 100 мл. Прибавляют 3 - 4 капли раствора фенолфталеина, и если окажется, что проба имеет щелочную реакцию, ее нейтрализуют серной кислотой до исчезновения окраски и прибавляют сверх того еще 0,5 мл кислоты. Если проба не покажет щелочной реакции, ее подкисляют добавлением 0,5 мл серной кислоты. Раствор нагревают до 40 - 50 °C и прибавляют 0,5 мл раствора железоаммонийных квасцов. В присутствии гексацианоферрата образуется осадок берлинской лазури. В этом случае пробу фильтруют через плотный стеклянный фильтр.
Раствор, окрашенный в красный цвет, подогревают до 40 - 50 °C и обесцвечивают добавлением 0,5 мл раствора пиросульфита. Смесь доводят до медленного кипения и прибавляют к ней 5 мл раствора сульфата меди. Выпадает роданид меди, образующий тонкий слой осадка, превращающийся при перемешивании в хлопья. Смесь оставляют на 15 мин в покое и потом в горячем состоянии фильтруют через плотный фильтр (синяя лента). Осадок на фильтре промывают горячей водой до тех пор, пока промывные воды не перестанут показывать с гексацианоферратом реакцию на медь. Фильтр прокалывают палочкой и смывают осадок дистиллированной водой в стакан. Фильтр промывают сначала 25 мл раствора карбоната натрия, потом горячей водой и оставляют для дальнейшей обработки. Промывные воды присоединяют к осадку в стакане.
Щелочной раствор с осадком нагревают до кипения, при этом роданид меди разлагается. Для лучшей коагуляции осажденных окисей меди можно добавить несколько капель раствора железоаммонийных квасцов. Горячий раствор фильтруют через плотный фильтр (синяя лента) и осадок основательно промывают горячей дистиллированной водой. Сохраненный ранее фильтр, обработанный раствором карбоната натрия, помещают в стакан, добавляют к нему 25 мл раствора карбоната натрия, размельчают фильтр палочкой и смесь короткое время кипятят. Потом раствор фильтруют через фильтр, которым пользовались при отделении окисей меди. Фильтраты соединяют, подкисляют их добавлением 10 мл раствора азотной кислоты и перемешивают. По охлаждении прибавляют 1,5 мл раствора железоаммонийных квасцов и красный раствор титруют 0,05 н. раствором нитрата серебра до обесцвечивания.
Расчет. Содержание роданид-ионов (x) в мг/л вычисляют по формуле

где a - объем 0,05 н. раствора нитрата серебра, израсходованного на титрование, мл;
k - поправка для приведения нормальности раствора нитрата серебра к точно 0,05 н.;
V - объем пробы, взятой для определения, мл.
Округление результатов
Диапазон, мг/л | 10 - 50 | 50 - 100 | 100 - 200 | 200 - 500 |
Округление, мг/л | 1 | 5 | 10 | 20 |
Колориметрический метод определения с бензидином и пиридином
38.7. Определение основано на превращении роданид-ионов (вместе с цианид-ионами) в бромистый циан, который реагирует с бензидином и пиридином с образованием полиметинового красителя.
Если окраска анализируемой сточной воды или ее мутность мешают определению, то полученный краситель экстрагируют н-амиловым или н-бутиловым спиртом и определяют его колориметрически в экстракте по варианту Б.
Описанным методом можно, не разбавляя пробу, определить роданид-ионы в концентрациях от 0,05 до 3 мг/л (вариант А) или 0,01 до 1,00 мг/л (вариант Б).
Мешающие влияния
38.8. Прямое колориметрическое определение роданидов нельзя провести, если вода содержит также цианиды. В этом случае цианиды предварительно или удаляют отгонкой в виде цианистого водорода, или разрушают их добавлением формальдегида: 2 мл анализируемого раствора, содержащего роданид в концентрациях от 0,05 до 3 мг/л, помещают в градуированный цилиндр, снабженный притертой пробкой. Добавляют 1 каплю 1%-ного раствора формалина, закрывают пробкой, перемешивают и дают постоять 5 мин. После этого вносят бромную воду до появления избытка брома (слегка желтое окрашивание) и снова дают постоять 5 - 10 мин, чтобы весь избыток формалина успел окислиться. Далее, уничтожив избыток брома прибавлением раствора мышьяковистой кислоты или гидразина, обрабатывают пробу пиридин-бензидиновым реактивом и измеряют оптическую плотность окраски возникшего полиметинового красителя по варианту А или Б.
Присутствие небольших количеств окислителей и восстановителей не мешает определению, поскольку в ходе работы сточную воду поочередно окисляют бромной водой и восстанавливают мышьяковистой кислотой. Наличие больших количеств этих веществ и сероводорода, а также ароматических аминов и красителей мешает определению. Сульфид-ионы нужно удалить прибавлением соли свинца.
Аппаратура
38.9. Градуированные стеклянные цилиндры или плоскодонные пробирки с притертой пробкой и меткой, обозначающей объем 2 мл.
Водяная баня.
Фотометр, кюветы длиной 1 см (вариант А), светофильтр  ; кюветы длиной 1 см (вариант Б), светофильтр
; кюветы длиной 1 см (вариант Б), светофильтр  .
.
; кюветы длиной 1 см (вариант Б), светофильтр .Реактивы
38.10. Бромная вода. Дистиллированную воду насыщают бромом ч.д.а.
Мышьяковистая кислота, 2%-ный раствор: 1,6 г трехокиси мышьяка As2O3 ч.д.а. растворяют в 100 мл воды при кипячении с обратным холодильником.
Гидразин солянокислый или гидразин сернокислый 0,5%-ный раствор.
Пиридин + соляная кислота, пиридиновый реактив: 60 мл свежеперегнанного пиридина (темп. кип. 114 °C) смешивают с 40 мл воды и 10 мл концентрированной соляной кислоты ч.д.а.
Бензидин солянокислый, 5%-ный раствор в разбавленной соляной кислоте: 5 г бензидина ч.д.а. растворяют в 100 мл разбавленной (2:98) HCl ч.д.а.
Метиловый оранжевый, индикатор (приготовление см. стр. 36).
Соляная кислота, 0,1 н. раствор (приготовление см. стр. 42).
Роданид калия, стандартный раствор:
а) запасной: 0,1673 г ч.д.а. растворяют в дистиллированной воде и разбавляют водой до 1 л; 1 мл раствора содержит 0,100 мг CNS-;
б) рабочий I: 30,0 мл запасного раствора разбавляют до 1 л дистиллированной водой. Применяют всегда свежеприготовленный раствор; 1 мл содержит 0,003 мг CNS-;
в) рабочий II: 10,0 мл запасного стандартного раствора разбавляют до 1 л дистиллированной водой; 1 мл раствора содержит 0,001 мл CNS-. Применяют всегда свежеприготовленный раствор.
н-Амиловый или н-бутиловый спирт ч.д.а. применяется в варианте Б.
Формальдегид, 1%-ный раствор.
Ход определения
38.11. Вариант А (определение в прозрачных и бесцветных водах).
В градуированный цилиндр или пробирку с притертой пробкой отмеряют 2 мл первоначальной пробы или предварительно разбавленной, чтобы содержание роданидов в ней не превышало 0,08 мг. Раствор подкисляют таким количеством 0,1 н. соляной кислоты, чтобы после нейтрализации был прибавлен избыток ее в 0,05 - 0,06 мл.
Требуемое количество кислоты определяют титрованием другой пробы анализируемой воды с применением метилового оранжевого в качестве индикатора.
Открытый цилиндр помещают на 30 мин в кипящую водяную баню. После этого содержимое цилиндра охлаждают и доводят объем смеси дистиллированной водой до 2 мл. Прибавляют 0,2 мл бромной воды и перемешивают. Избыток брома удаляют, добавляя 0,2 мл 2%-ного раствора мышьяковистой кислоты. (Вместо раствора мышьяковистой кислоты можно применить 0,5%-ный раствор солянокислого или сернокислого гидразина, который прибавляют по каплям до исчезновения окраски брома и сверх того еще 1 каплю). Большого избытка следует избегать; обычно расходуется 0,25 - 0,35 мл указанного раствора.) Содержимое цилиндра хорошо перемешивают. Отдельно приготовляют в пробирке раствор бензидин-пиридинового реактива смешением 3 мл раствора пиридина и 0,6 мл раствора бензидина. Приготовленную смесь приливают к пробе в цилиндре и все перемешивают повторным переливанием из одной посуды в другую. Через 15 - 20 мин измеряют оптическую плотность, вычитают значение ее для холостого опыта, проведенного таким же образом с 2 мл дистиллированной воды, и по калибровочной кривой находят содержание роданид-ионов.
Калибровочная кривая
38.12. В ряд градуированных цилиндров отмеряют 0 - 0,10 - 0,20 - 0,50 - 1,00 - 1,50 - 2,00 мл рабочего стандартного раствора I и разбавляют каждый раствор дистиллированной водой до 2 мл; в результате получают концентрации 0 - 0,15 - 0,30 - ... - 3,0 мг роданид-ионов в 1 л. Эти растворы обрабатывают описанным выше методом, измеряют оптическую плотность, вводят поправку на холостое определение с дистиллированной водой и строят график зависимости оптической плотности от концентрации роданид-ионов.
Вариант Б (определение в мутных и цветных водах). Для определения берут 5 мл первоначальной пробы или предварительно разбавленной, чтобы в этом объеме содержалось 0,05 - 5 мкг роданидов. Продолжают, как описано в варианте А, но после удаления избытка брома прибавляют к смеси 5,0 мл н-бутилового или н-амилового спирта, закрывают цилиндр пробкой и взбалтывают. После этого приливают бензидин-пиридиновый реактив, как в варианте А, закрывают пробкой, сильно взбалтывают и дают постоять не менее 15 мин. Слой органического растворителя переводят в кювету фотометра и измеряют оптическую плотность, вводят поправку на холостое определение, проведенное таким же способом с 5 мл дистиллированной воды.
Калибровочная кривая
38.13. В ряд градуированных цилиндров отмеряют 0 - 0,10 - 0,50 - 1,00 - 2,00 - 3,00 и 5,00 мл рабочего раствора II и разбавляют каждый раствор до 5 мл; в результате получают концентрации 0 - 0,1 - 0,5 - ... - 5,0 мг роданид-ионов в 1 л. Эти растворы обрабатывают приведенными выше методами. Калибровочную кривую строят так же, как в варианте А; для холостого определения берут один из указанных растворителей.
Расчет. Содержание роданид-ионов в мг/л, определенное по варианту А (x) или Б (y), вычисляют по формулам:
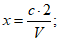

где c - концентрация роданидов, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для анализа, мл.
Округление результатов
Диапазон, мг/л | 0,050 - 0,100 | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,00 |
Округление, мг/л | 0,005 | 0,01 | 0,02 | 0,05 |
Общие положения
39.1. Соединения кремниевой кислоты попадают в природные воды вследствие выщелачивания их из горных пород водой, содержащей углекислый газ. Встречаются они в водах в виде окиси, в той или иной степени гидратированной, в виде алюмосиликатов, а также в ионизированной форме в виде ортосиликат-ионов. Преобладание ионизированной или неионизированной формы зависит от величины pH воды.
Обычно концентрация кремнекислых соединений в природных поверхностных водах не превышает 10% их общего солесодержания. В подземных водах концентрация кремнекислоты может быть значительно выше, достигая 25 - 30% величины их солесодержания.
Для определения кремнекислоты в естественных водах предлагаются: весовой метод и колориметрический метод, основанный на образовании кремнемолибденового комплексного соединения, окрашенного в желтый цвет.
Весовым методом пользуются для определения в воде всех растворенных силикатов, а также для определения концентрации кремнекислоты в стандартных растворах кремнекислого натрия, применяемых в колориметрическом методе. Метод удобен для воды, содержащей не менее 20 мг/л SiO2.
Колориметрический метод рекомендуется для анализа прозрачных и слегка мутных вод, содержащих от 0,4 до 25 мг/л SiO2. Этот интервал может быть увеличен разбавлением исходной воды. Колориметрическим методом можно определить растворенные ортосиликаты, а также все растворенные силикаты реакцией с молибдатом после гидролиза в щелочной среде.
Пробы воды не консервируют; отбор производят в сосуды из полиэтилена; менее желательна посуда из химически устойчивого стекла, особенно если неизбежен промежуток между отбором пробы и анализом.
Весовое определение всех силикатов
39.2. В результате выпаривания пробы воды досуха с добавлением соляной кислоты образуется кремнекислота в нерастворимой форме, которую отфильтровывают, высушивают, прокаливают и взвешивают в виде окиси кремния. Весовым методом можно определить не менее 5 мг SiO2 в объеме пробы, взятой для анализа.
Точность метода ограничивается как растворимостью SiO2 в воде, так и соблюдением всех мер предосторожности при отборе проб и проведении анализа. Точность приближается к +/- 0,2 мг/л SiO2. Если для анализа берется 1 л пробы, точность возрастает до 0,02 мг/л SiO2.
Мешающие влияния
39.3. Определению мешает высокое содержание органических веществ. Их удаляют минерализацией. Профильтрованную пробу выпаривают в платиновой чашке досуха после добавления 1 мл концентрированной H2SO4 и 3 мл концентрированной HNO3 и после исчезновения дыма слегка прокаливают. Затем приливают 50 мл дистиллированной воды и 5 мл HCl и продолжают обработку пробы, как указано ниже. Определению мешают фториды.
Аппаратура
39.4. Платиновая чашка емкостью 200 мл.
Платиновый тигель с крышкой.
Водяная баня.
Муфельная или тигельная печь (1200 °C).
Реактивы
39.5. Соляная кислота ч.д.а., концентрированная.
Серная кислота ч.д.а., концентрированная.
Фтористоводородная кислота ч.д.а., концентрированная.
Ход определения
39.6. Отбирают от 200 до 100 мл профильтрованной пробы. Прибавляют 5 мл концентрированной соляной кислоты и выпаривают постепенно на водяной бане досуха. Обработку остатка соляной кислотой повторяют еще два раза, затем просушивают в течение 1 ч при 110 °C. По охлаждении наливают в чашку 5 мл HCl, нагревают и прибавляют примерно 50 мл горячей дистиллированной воды. Суспензию фильтруют через беззольный фильтр средней плотности (белая лента). Чашку и осадок промывают горячей HCl (1:50) и затем минимальным объемом дистиллированной воды до исчезновения хлоридов в промывных водах.
Фильтр с остатком переносят в платиновый тигель, высушивают при 110 °C и прокаливают при 1200 °C до постоянного веса. Остаток после прокаливания смачивают несколькими каплями концентрированной серной кислоты и прибавляют 10 мл фтористоводородной кислоты. Смесь выпаривают досуха на водяной бане и прокаливают при 1200 °C.
Отдельно определяют содержание посторонних примесей в 10 мл фтористоводородной кислоты путем выпаривания, прокаливания до постоянного веса и взвешивания, результаты вычитают из веса тигля при втором взвешивании.
Расчет. Общее содержание растворенных силикатов SiO2 (x) в мг/л вычисляют по формуле

где a - вес тигля с окисью кремния, мг;
a1 - вес тигля после обработки осадка фтористоводородной кислотой, мг;
V - объем пробы, взятой для определения, мл.
Колориметрическое определение кремнекислоты с молибдатом
39.7. Определение растворенных ортосиликатов
Растворенные ортосиликаты дают с молибдатом в кислой среде окрашенную в желтый цвет комплексную гетерополикислоту H2[Si(Mo3O10)4]·H2O, пригодную для колориметрии.
Определение концентрации кремнекислоты визуально в цилиндрах Несслера дает точность +/- 5%. Фотометрическое измерение дает точность 1% и более.
Мешающие влияния
39.8. Определению мешают таннин, большие количества железа, цветность, мутность, сульфиды и фосфаты. Обработка пробы щавелевой кислотой устраняет мешающее влияние фосфатов и снимает действие таннина. Чтобы устранить влияние окраски самой пробы или мутности ее, сохранившейся после фильтрования, вычитают оптическую плотность подкисленной пробы без добавления молибдата из найденного опытом значения оптической плотности пробы воды после выполнения хода определения.
Мешающее влияние сероводорода устраняют продуванием пробы воздухом. Сильно щелочные пробы нейтрализуют разбавленной серной кислотой до  , используя в качестве индикатора п-динитрофенол.
, используя в качестве индикатора п-динитрофенол.
, используя в качестве индикатора п-динитрофенол.Этим методом можно определить от 1 до 20 мг/л SiO2 с точностью +/- 0,5 мг/л.
Аппаратура
39.9. Фотометр, светофильтр  .
.
.Кюветы, толщиной 2 - 5 см или набор цилиндров Несслера емкостью 50 мл.
Мерные колбы емкостью 50 мл.
Реактивы
39.10. Молибдат аммония, 10%-ный раствор: растворяют 10 г (NH4)6Mo7O24·4H2O в слегка подогретой дистиллированной воде при перемешивании и разбавляют до 100 мл. Если нужно, фильтруют. Раствор молибдата аммония сохраняется продолжительное время, если pH довести до 7 - 8 свободным от кремнекислоты раствором аммиака или NaOH и хранить в полиэтиленовой посуде.
Соляная кислота ч.д.а., разбавленная (1:1).
Щавелевая кислота, 10%-ный раствор: 10 г H2C2O4·2H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 100 мл.
Силикат натрия, стандартный раствор:
а) запасной: растворяют 4,73 г Na2SiO3 ч.д.а. в свежепрокипяченной и охлажденной дистиллированной воде и доводят приблизительно до 900 мл. Содержание SiO2 в этом растворе определяют весовым методом. В зависимости от полученного результата раствор разбавляют так, чтобы 1 мл его содержал 1 мг SiO2;
б) рабочий: 50 мл запасного стандартного раствора разбавляют свежепрокипяченной и охлажденной дистиллированной водой до 1 л.
Следует всегда применять свежеприготовленный раствор; 1 мл раствора содержит 0,05 мг SiO2. Стандартные растворы хранят в полиэтиленовых или парафинированных сосудах.
Хромат калия, стандартный раствор для сравнения: 0,252 г K2CrO4 ч.д.а. растворяют в дистиллированной воде и разбавляют до 200 мл; 1 мл этого раствора, разбавленный до 55 мл, дает окраску, соответствующую 4 мг/л SiO2.
Бура, 5%-ный раствор: 10 г Na2B4O7·10H2O ч.д.а. растворяют в дистиллированной воде и разбавляют до 200 мл.
Калибровочная кривая
39.11. В несколько колб из устойчивого стекла наливают 0 - 1,0 - 2,0 до 20 мл рабочего раствора и доливают дистиллированной водой до 50 мл. Приготовленные таким образом растворы содержат 0 - 1,0 - 2,0 до 20 мг SiO2 в 1 л. Растворы обрабатывают описанным ниже способом. Измеряют оптическую плотность и, вычитая значение ее для холостого определения, строят график зависимости оптической плотности от концентрации SiO2. При подготовке стандартных растворов в цилиндрах Несслера ход работы такой же, как при построении калибровочной кривой. Шкалу растворов получают введением в цилиндры Несслера 0 - 0,25 - 0,5 до 5 мл раствора хромата, прибавлением в каждый раствор 5 мл раствора буры и разбавлением дистиллированной водой до 55 мл. Стандартные растворы соответствуют по своей окраске содержанию 0 - 1,0 - 2,0 до 20 мг/л SiO2.
Ход определения
39.12. В колбу из устойчивого стекла наливают 50 мл прозрачной пробы или меньший объем пробы и разбавляют ее дистиллированной водой до 50 мл. Прибавляют 2 мл раствора молибдата и перемешивают. Затем приливают 1 мл соляной кислоты и смесь снова перемешивают. Через 5 мин прибавляют 1,5 мл раствора щавелевой кислоты и измеряют оптическую плотность или сравнивают окраску со стандартной шкалой в цилиндрах Несслера или, наконец, определяют SiO2 колориметрическим титрованием. Одновременно проводят холостое определение с дистиллированной водой, вводят поправку и по калибровочной кривой находят содержание SiO2.
Для определения SiO2 колориметрическим титрованием берут новую колбу такой же формы и емкости. В нее наливают 5 мл раствора буры и 50 мл дистиллированной воды. Затем прибавляют из бюретки раствор хромата до тех пор, пока цвет раствора в этой колбе не совпадет с окраской пробы. В колбу с пробой доливают столько дистиллированной воды, сколько добавили раствора хромата в колбу для сравнения. Объем израсходованного раствора хромата в миллилитрах умножают на 4 и получают значение c для расчета по приведенной ниже формуле.
Расчет. Содержание растворенных ортосиликатов (x) в мг/л вычисляют по формуле

где c - концентрация SiO2, найденная по калибровочной кривой сравнением со стандартами или колориметрическим титрованием, мг/л;
V - объем анализируемой пробы, мл.
Определение всех растворенных силикатов
39.13. Все формы ортосиликатов в растворе превращают щелочным гидролизом в ортоформу, в которой их и определяют. В нейтрализованной пробе колориметрическим методом определяют концентрацию растворенных ортосиликатов.
Мешающие влияния
39.14. Мешают те же факторы, что и при определении растворенных ортосиликатов.
Аппаратура
39.15. Платиновая чашка, обработанная 20%-ным раствором едкого натра при кипячении.
Реактивы
39.16. Бикарбонат натрия ч.д.а.
Едкий натр, 20%-ный раствор (хранить в полиэтиленовом сосуде).
Серная кислота, ч.д.а., 1 н. раствор: осторожно приливают 28 мл концентрированной H2SO4 к 800 мл дистиллированной воды при перемешивании, охлаждают и разбавляют до 1 л.
Другие реактивы, необходимые для колориметрического определения (см. стр. 196).
Ход определения
39.17. К 50 мл профильтрованной пробы прибавляют в платиновой чашке 0,2 г NaHCO3 или 1 мл 20%-ного раствора едкого натра. Смесь нагревают на кипящей водяной бане 1 ч, если прибавили NaHCO3 и 20 мин при прибавлении NaOH. По охлаждении для нейтрализации раствора прибавляют при помешивании 2,4 мл H2SO4. После этого сразу переходят к дальнейшим операциям. Раствор количественно переносят в мерную колбу емкостью 50 мл и разбавляют дистиллированной водой до метки. Таким же образом обрабатывают раствор в холостом опыте с дистиллированной водой. Далее метод не отличается от колориметрического определения растворенных ортосиликатов (см. стр. 195).
Расчет. Содержание всех растворенных силикатов рассчитывают по формуле, приведенной для определения растворенных ортосиликатов (см. стр. 197).
Общие положения
40.1. Естественное содержание сульфатов в поверхностных и грунтовых водах обусловлено выветриванием пород и биологическими процессами в водоносных слоях. Содержание сульфатов повышается в водоемах вследствие сброса в них сточных вод с неорганическими и органическими соединениями серы.
В питьевых и поверхностных водах сульфаты определяют комплексонометрическим титрованием (содержание сульфатов до 1000 мг/л) или весовым методом в виде сульфата бария.
Пробы обычно не консервируют. Результаты приводятся в миллиграмм-эквивалентах или миллиграммах сульфатов на 1 л воды:  ;
;  .
.
; .Качественное определение
40.2. Примерно 10 мл пробы подкисляют в пробирке несколькими каплями соляной кислоты и прибавляют около 0,5 мл 10%-ного раствора хлорида бария. При содержании 5 - 50 мг/л сульфатов возникает опалесценция или слабое помутнение, при более высоком содержании выпадает осадок.
Комплексонометрическое определение
40.3. Пробу воды, пропущенную через катионит в Н-форме, осаждают титрованным раствором хлорида бария. Избыток ионов бария определяется комплексонометрическим титрованием титрованным раствором комплексона III в аммиачной среде с применением крезолфталексона в качестве индикатора. Обратным титрованием раствором хлорида бария определяют точку эквивалентности.
Точность определения +/- 0,5 мг/л при титровании 250 мл пробы с содержанием 2 - 50 мг сульфатов в 1 л воды.
Мешающие влияния
40.4. Определению мешают катионы, которые реагируют с раствором комплексона III. Катионы должны быть удалены полностью, поэтому работу применяемой ионообменной колонки надо постоянно контролировать.
Взвешенные вещества засоряют катионитную колонку, мутность мешает определению точки эквивалентности. Аналогичное влияние оказывает также повышенное содержание железа (свыше 5 мг/л). Взвешенные вещества и мутность устраняются предварительным фильтрованием или центрифугированием пробы. При повышенном содержании железа его осаждают разбавленным раствором аммиака. К насыщенной воздухом пробе прибавляют разбавленный (1:10) раствор аммиака до щелочной реакции и кипятят до исчезновения аммиачного запаха. Выделившуюся гидроокись железа отфильтровывают, осадок на фильтре промывают дистиллированной водой, и фильтрат доводят до первоначального объема.
Аппаратура
40.5. Колонка с катионитом в Н-форме.
Н-катионитовая колонка, служащая для замещения всех катионов растворенных в воде солей на катион водорода, представляет собой стеклянную трубку диаметром 18 - 25 мм, высотой не менее 70 см; нижний конец трубки оттянут на конус, и в него впаян кран, степенью открытия которого можно регулировать скорость течения жидкости через колонку. Над конической частью в трубку впаяно стеклянное пористое дно. Допускается замена пористого дна стеклянной ватой, заполняющей нижнюю часть трубки на высоту 25 - 30 мм.
Стеклянная трубка устанавливается вертикально в штативе и заполняется на высоту 40 - 45 см отмытым кислотой катионитом Ку-2 или сульфоуглем (сорт 1, крупный). Отмывка катионита или сульфоугля до загрузки в колонку кислотой осуществляется следующим образом. Навеска в 100 г катионита помещается в стеклянный или фарфоровый стакан, заливается 500 мл 5%-ного раствора HCl в дистиллированной воде. Смесь перемешивается стеклянной палочкой и оставляется на 4 ч. По прошествии этого времени раствор кислоты заменяется свежим, смену растворов производят 5 раз с интервалами 4 ч, затем катионит переносят в стеклянную трубку, предварительно на 1/3 заполненную 5%-ным раствором кислоты. Количество катионита, перенесенного в трубку, должно образовывать в ней слой высотой 40 см. Затем через слой катионита медленно (1 мл в минуту) пропускают в течение 6 ч 5%-ный раствор кислоты и затем отмывают катионит от кислоты до нейтральной по метиловому оранжевому реакции дистиллированной водой.
Реактивы
40.6. Хлорид бария, 0,05 М раствор: 12,214 г BaCl22H2O ч.д.а. растворяют в дистиллированной воде, подкисляют 1 мл концентрированной HCl ч.д.а. и доводят до 1 л. Титр или поправку раствора определяют весовым путем осаждением в виде сульфата бария. Берут средний из трех определений.
Комплексон III, 0,05 М раствор: 18,6 г комплексона III растворяют в дистиллированной воде и разбавляют до 1 л. Титр или поправку раствора определяют титрованием 0,05 М точным раствором хлорида бария. К 100 мл дважды дистиллированной воды в колбе для титрования прибавляют 5 мл 0,25 М титрованного раствора хлорида бария с известной поправкой. Затем прибавляют 5 мл концентрированного аммиака, 0,1 мл раствора индикатора и около 20 мл спирта. Фиолетовый раствор титруют раствором комплексона III до обесцвечивания. Точку эквивалентности определяют обратным титрованием раствором хлорида бария. Титр или поправку раствора комплексона III определяют по среднему значению результатов трех титрований.
Аммиак ч.д.а., концентрированный.
Крезолфталексон, 1%-ный раствор: 0,05 г крезолфталексона (2,2'-диацетиламинометилкрезолфталеин) растворяют в 5 мл концентрированного раствора аммиака. Раствор устойчив около 8 ч.
Этиловый спирт, 96%-ный, нельзя применять спирт, денатурированный примесью, вызывающей при определении помутнение раствора.
Ход определения
40.7. Пробу воды медленно пропускают через колонку с катионитом в Н-форме. Первые 30 - 50 мл пробы отбрасывают, из следующей части отбирают в колбу для титрования емкостью 500 мл такое количество пробы, чтобы в титруемой жидкости содержалось 1 - 10 мг сульфат-ионов (при предполагаемом содержании сульфатов от 1 до 50 мг/л отбирают 250 мл, при концентрациях от 50 до 100 мг/л - 100 мл и т.д.).
К отобранной пробе, объем которой доводят дистиллированной водой до 200 - 250 мл, прибавляют из микробюретки точно 4 мл раствора хлорида бария. Смесь кипятят 2 - 5 мин и оставляют не менее чем на 5 ч.
Затем прибавляют около 10 мл концентрированного раствора аммиака, 0,2 мл раствора индикатора и около 30 мл спирта. Титруют из микробюретки с делениями через 0,02 мл раствором комплексона III до исчезновения красно-фиолетовой окраски. Если индикатора прибавили больше, чем указано выше (или если качество индикатора недостаточно высокое), то может остаться слабая розовая окраска. Записывают расход раствора комплексона III. Затем вторично титруют раствором хлорида бария до момента начала изменения окраски из бесцветной в красно-фиолетовую и записывают расход раствора хлорида бария. При объеме 250 мл можно заметить изменение окраски уже после прибавления 0,04 мл титрованных растворов.
Расчет. Содержание сульфат-ионов в мг/л (x) или мг-экв/л (y) вычисляют по формулам:

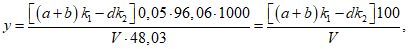
где a - прибавленный объем 0,05 М раствора хлорида бария, мл;
b - расход 0,05 М раствора хлорида бария при вторичном титровании, мл;
d - расход 0,05 М раствора комплексона, мл;
k1 - поправка для приведения концентрации раствора хлорида бария к точно 0,05 М;
k2 - поправка для приведения концентрации раствора комплексона III к точно 0,05 М;
96,06 - молекулярный вес  ;
;
;48,03 - эквивалент  .
.
.Округление результатов
Диапазон, мг/л | 2,0 - 5,0 | 5,0 - 10,0 | 10,0 - 20,0 | 20,0 - 50,0 и т.д. |
Округление: | ||||
мг/л | 0,5 | 1 | 2 | 5 |
мг-экв/л | 0,01 | 0,02 | 0,05 | 0,1 |
Весовое определение
40.8. Малая растворимость сульфата бария (произведение растворимости равно 1,98·10-10) позволяет производить весовое определение сульфат-ионов в слабокислой среде. Подходящим объемом пробы является 250 мл с содержанием  - 100 мг/л. Точность определения +/- 2 мг
- 100 мг/л. Точность определения +/- 2 мг  на 1 л воды.
на 1 л воды.
Мешающие влияния
40.9. Определению мешают высокое содержание силикатов и железа, взвешенные и коллоидные вещества. Одновременно с сульфатами определяются и сульфиты. Взвешенные и коллоидные вещества устраняют фильтрованием или центрифугированием.
Высокое содержание кремнекислоты и железа устраняют выпариванием взятого объема пробы досуха после подкисления 5 мл HCl (1:1). Сухой остаток подсушивают около 1 ч при температуре 105 °C, смачивают 5 мл HCl (1:1), нагревают и разбавляют примерно до 50 мл дистиллированной водой. Горячий раствор фильтруют, промывают разбавленной (1:50) соляной кислотой. К фильтрату прибавляют 1 мл азотной кислоты (1:1), слабо кипятят и в некоторых случаях упариванием доводят объем примерно до 100 мл. Затем прибавляют разбавленный (1:1) раствор аммиака до явно щелочной реакции и смесь нагревают на водяной бане примерно полчаса. Выделившиеся гидроокиси отфильтровывают и промывают горячей дистиллированной водой. Объем фильтрата доводят примерно до 250 мл.
В присутствии сульфитов пробу предварительно окисляют добавлением необходимого количества 0,1 н. раствора йода. Из результатов определения сульфатов вычитают найденное отдельно содержание сульфитов.
Аппаратура
40.10. Водяная баня.
Муфельная или тигельная печь (800 °C).
Реактивы
40.11. Соляная кислота ч.д.а., разбавленная (1:1).
Хлорид бария, 10%-ный раствор, для осаждения: 10 г BaCl2·2H2O ч.д.а. растворяют в дистиллированной воде и доводят до 100 мл.
Нитрат серебра, 1,7%-ный раствор: 8,5 г AgNO3 ч.д.а. растворяют в 500 мл дистиллированной воды и подкисляют 0,5 мл концентрированной HNO3 ч.д.а.
Ход определения
40.12. К 250 мл пробы с содержанием 5 - 50 мг  или же к меньшему объему с таким же содержанием сульфатов, но доведенному до 250 мл дистиллированной водой, помещенной в стакан емкостью 400 - 600 мл, прибавляют 2 мл разбавленной соляной кислоты. Смесь нагревают до кипения и при постоянном перемешивании прибавляют 3 мл горячего раствора хлорида бария. Смесь перемешивают около 1 мин, нагревают в течение 1 ч на водяной бане и оставляют на 8 - 12 ч при комнатной температуре. Фильтруют через плотный фильтр (синяя лента) и промывают декантацией. Выделившийся сульфат бария переводят количественно на фильтр. Приставшие к стенкам стакана следы сульфата бария удаляют обтиранием влажным кусочком беззольной фильтровальной бумаги и ополаскивают стакан дистиллированной водой. Осадок на фильтре промывают горячей дистиллированной водой до отсутствия хлоридов в фильтрате, определяемых качественно раствором нитрата серебра. Фильтр с осадком переносят во взвешенный предварительно прокаленный тигель. После высушивания фильтр осторожно сжигают и осадок прокаливают при 800 °C до постоянного веса и взвешивают BaSO4.
или же к меньшему объему с таким же содержанием сульфатов, но доведенному до 250 мл дистиллированной водой, помещенной в стакан емкостью 400 - 600 мл, прибавляют 2 мл разбавленной соляной кислоты. Смесь нагревают до кипения и при постоянном перемешивании прибавляют 3 мл горячего раствора хлорида бария. Смесь перемешивают около 1 мин, нагревают в течение 1 ч на водяной бане и оставляют на 8 - 12 ч при комнатной температуре. Фильтруют через плотный фильтр (синяя лента) и промывают декантацией. Выделившийся сульфат бария переводят количественно на фильтр. Приставшие к стенкам стакана следы сульфата бария удаляют обтиранием влажным кусочком беззольной фильтровальной бумаги и ополаскивают стакан дистиллированной водой. Осадок на фильтре промывают горячей дистиллированной водой до отсутствия хлоридов в фильтрате, определяемых качественно раствором нитрата серебра. Фильтр с осадком переносят во взвешенный предварительно прокаленный тигель. После высушивания фильтр осторожно сжигают и осадок прокаливают при 800 °C до постоянного веса и взвешивают BaSO4.
Расчет. Содержание сульфат-ионов вычисляют в мг/л (x) или в мг-экв/л (y) по формулам:


где m - вес BaSO4, мг;
V - объем пробы, взятой для определения, мл;
0,4116 - коэффициент для пересчета с BaSO4 на  ;
;
48,03 - эквивалент  .
.
.Округление результатов
Диапазон, мг/л | 20 - 100 | 100 - 200 | 200 - 500 | 500 - 1000 |
Округление: | ||||
мг/л | 1 | 2 | 5 | 10 |
мг-экв/л | 0,02 | 0,05 | 0,1 | 0,2 |
Определение сульфатов объемным методом титрованием раствором
нитрата свинца
40.13. Сульфаты определяются прямым титрованием в щелочной среде раствором нитрата свинца с применением кристаллического дитизона в качестве индикатора.
Метод чувствительный и точный, применяется для определения сульфатов в природных водах.
Мешающие влияния
40.14. Все катионы, которые оказывают мешающие влияния, устраняются пропусканием пробы через катионит в Н-форме. Из анионов мешающее влияние оказывают фосфаты. Определению могут мешать и другие анионы, которые осаждаются свинцом, например хромат-, арсенат-, фторид-, йодид- и оксалат-ионы, которые в природных водах не встречаются или содержатся в микрограммовых количествах.
Аппаратура
40.15. Колонка с катионитом, в Н-форме.
Реактивы
40.16. Нитрат свинца 0,02 н. раствор 3,31 г Pb(NO3)2 растворяют в 1 л воды, титр этого раствора устанавливают по стандартному 0,02 н. раствору сульфата натрия.
Дитизон, кристаллический дитизон растирают с бензойной кислотой (1:50) в порошок.
Этиловый спирт, 96%-ный, чистый.
Соляная кислота (1:3) для регенерации ионита.
Ход определения
40.17. Пробу воды пропускают через колонку с катионитом в Н-форме для устранения мешающих определению катионов.
Отбирают такое количество элюата, чтобы в объеме 10 - 20 мл содержалось не меньше 1 мг  . Если сульфатов содержится мало (10 - 20 мг/л
. Если сульфатов содержится мало (10 - 20 мг/л  ), то отбирают 50 мл или больше в коническую колбу и раствор упаривают приблизительно до 10 - 20 мл. Прибавляют двойное количество спирта (т.е. 20 - 40 мл) и порошкообразный дитизон в таком количестве, чтобы раствор имел зеленую окраску, затем титруют из микробюретки раствором нитрата свинца до перехода зеленой окраски в красно-фиолетовую. В случае низкой концентрации сульфатов применяют более разбавленные растворы нитрата свинца.
), то отбирают 50 мл или больше в коническую колбу и раствор упаривают приблизительно до 10 - 20 мл. Прибавляют двойное количество спирта (т.е. 20 - 40 мл) и порошкообразный дитизон в таком количестве, чтобы раствор имел зеленую окраску, затем титруют из микробюретки раствором нитрата свинца до перехода зеленой окраски в красно-фиолетовую. В случае низкой концентрации сульфатов применяют более разбавленные растворы нитрата свинца.
Расчет. Содержание сульфат-ионов (x) в мг/л вычисляют по формуле

где a - расход 0,02 н. раствора нитрата свинца на титрование, мл;
k - поправка для приведения нормальности раствора нитрата свинца к точно 0,02 н.;
V - объем пробы, взятой для определения, мл;
48,03 - эквивалент  .
.
Общие положения
41.1. Сероводород как в свободном состоянии, так и в виде солей сероводородной кислоты (сульфидов и гидросульфидов) встречается в поверхностных и в подземных водах, причем его содержание (в пересчете на H2S) нередко достигает нескольких десятков миллиграммов в 1 л воды.
Относительное содержание в воде сероводорода (H2S), гидросульфид-(HS-) и сульфид-ионов (S2-) зависит от величины pH воды и определяется расчетом при помощи констант диссоциации сероводородной кислоты. Ниже приведено содержание H2S в воде в зависимости от pH воды и общего количества соединений сероводорода при температуре 20 °C.
pH воды | 5 | 6 | 6,5 | 7,0 | 7,5 | 8 | 9 |
Содержание в воде H2S в % | 98 | 86 | 67 | 39 | 17 | 6 | 0,6 |
Приведенный ниже метод отличается высокой чувствительностью, а также простотой исполнения при выполнении серий анализов; допускает использование приборов для колориметрирования.
Пробы отбирают в склянки с притертой пробкой, обычно используемые для определения кислорода, и после добавления реактивов их можно хранить в темноте до 24 ч.
Колориметрическое определение
41.2. Метод основан на образовании зеленовато-голубого окрашивания при взаимодействии сероводорода с продуктами окисления этилоксиэтилпарафенилендиамина солями железа (III).
Вместо этилоксиэтилпарафенилендиамина можно применять также парафенилендиамин или диметилпарафенилендиамин.
Определению мешают Cu2+, а также 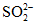 и
и  при содержании выше 40 мг/л, поэтому для разбавления используют дистиллированную воду, не содержащую следов перечисленных выше ионов.
при содержании выше 40 мг/л, поэтому для разбавления используют дистиллированную воду, не содержащую следов перечисленных выше ионов.
Во избежание большой относительной ошибки вследствие летучести сероводорода объем пробы для определения должен быть не меньше 25 мл.
Минимальная концентрация сероводорода, определяемая этим методом, 0,05 мг/л. Относительная погрешность определения в интервале концентраций 1,0 - 5,0 мг/л +/- 4%.
Аппаратура
41.3. Фотометр, светофильтр  .
.
.Склянки с притертой пробкой емкостью 100 - 200 мл.
Реактивы
41.4. Этилоксиэтилпарафенилендиамин, 0,3%-ный раствор: 0,3 г реактива растворяют в 100 мл 50%-ного раствора серной кислоты (х.ч.). Реактив сохраняется в холодильнике до двух месяцев.
Хлорное железо: 50 г FeCl3·6H2O (х.ч.) растворяют в дистиллированной воде, доводят объем до 50 мл; после отстаивания раствор сливают с осадка и хранят в холодильнике.
Сульфид натрия, стандартный раствор: 0,3 г Na2S·9H2O (ч.д.а.) растворяют в 150 мл дистиллированной воды и прибавляют 150 мл глицерина (х.ч.). Содержание сульфидов определяют йодометрическим титрованием (см. ниже) и затем разбавляют раствор так, чтобы 1 мл его содержал 0,1 мг H2S.
Титр приготовленного раствора устанавливают следующим образом. В склянку объемом 100 - 200 мл с притертой пробкой вносят 2,5 мл HCl (1:9), 50 мл 0,05 н. раствора йода и добавляют приготовленный раствор сульфида до верха склянки. После 10 мин выдерживания в темном месте титруют избыток йода 0,05 н. раствором тиосульфата. Аналогично ставят холостой опыт с йодом, прибавляя в склянку вместо раствора сульфида дистиллированную воду.
Общее содержание сероводорода в стандартном растворе (мг/л) вычисляют по формуле

где a - объем раствора тиосульфата, пошедший на титрование в холостом опыте, мл;
b - то же, пошедший на испытуемый раствор, мл;
k - поправочный коэффициент для приведения концентрации раствора тиосульфата к точно 0,05 н.;
0,85 - коэффициент пересчета на H2S;
V - объем склянки, мл.
Титр раствора тиосульфата (k) устанавливают по 0,05 н. раствору бихромата калия: в коническую колбу емкостью 0,5 л вносят 5 мл H2SO4 (1:3), прибавляют 1 - 2 г KJ и 10 мл 0,05 н. раствора K2Cr2O7. Колбу закрывают и оставляют стоять в темноте 5 мин. Затем добавляют 100 мл дистиллированной воды и титруют раствором тиосульфата с индикатором крахмалом до исчезновения окраски:

где n - объем раствора тиосульфата, пошедший на титрование, мл.
Тиосульфат натрия, 0,05 н. раствор.
Бихромат калия, 0,05 н. раствор.
Из приготовленного раствора Na2S с известным содержанием сероводорода разбавлением приготавливают эталонные растворы, содержащие от 0,10 до 1,0 мг/л сероводорода. Эти растворы используются для составления калибровочной кривой.
Ход определения
41.5. Пробы для определения сероводорода отбирают в склянки с притертой пробкой, обычно применяемые для определения кислорода (см. стр. 76), соблюдая те же предосторожности, как и при взятии пробы на кислород.
В склянку под уровень воды вводят 1 мл раствора этилоксиэтилпарафенилендиамина и после перемешивания 0,5 мл раствора хлорного железа (III). В течение 30 мин развивается зеленовато-голубое окрашивание. Определяют оптическую плотность и по калибровочной кривой вычисляют содержание сероводорода.
Если содержание сероводорода в воде превышает 1 мг/л, определение повторяют. При этом количество реактивов увеличивают таким образом, чтобы через 30 мин после введения реактивов и разбавления пробы дистиллированной водой до содержания сероводорода 0,7 - 0,8 мг/л концентрация реактивов в пробе была не менее указанной выше.
Расчет. Содержание сероводорода (x) в мг/л вычисляют по формуле
x = ac,
здесь c - найденное по калибровочной кривой количество сероводорода в пробе, мг/л;
a - кратность разбавления.
Общие положения
42.1. Сульфиты в поверхностных водах могут оказаться только в результате загрязнения их сточными водами.
Сульфиты определяются непосредственно йодометрически (вариант А) или удалением двуокиси серы из подкисленной пробы, поглощением ее раствором едкого натра и йодометрическим титрованием этого раствора (вариант Б). Прямое определение имеет преимущество при концентрации сульфитов, равной или превышающей 0,5 мг/л, и при отсутствии мешающих веществ. В противном случае применяется вариант Б. Колориметрически с фуксином сульфиты определяются после удаления двуокиси серы из подкисленной пробы и поглощения ее раствором едкого натра. Этим способом можно определять сульфиты при содержании 0,02 мг/л и более.
Так как сульфиты легко окисляются кислородом воздуха, определение проводится сразу же после отбора пробы или отобранную в склянку (кислородная склянка) пробу консервируют добавлением 0,2 мл 20%-ного раствора NaOH и 2,0 мл глицерина на каждые 100 мл. Консервированная проба должна быть проанализирована в течение суток.
Результаты выражаются в миллиграммах сульфит-ионов в 1 л воды.
Качественное определение
42.2. Приблизительно 50 мл пробы подкисляют в колбе с узким горлом несколькими каплями фосфорной кислоты. В горло предварительно вкладывают кусочек влажной йодокрахмальной бумаги.
В присутствии сульфитов сразу же после нагревания пробы бумага синеет. В случае большого количества сульфитов бумага через некоторое время снова обесцвечивается. Определению мешают тиосульфаты и сероводород.
Приготовление йодокрахмальной бумаги: 8 г йодата калия растворяют в 100 мл дистиллированной воды и этим раствором пропитывают полоски фильтровальной бумаги. Полоски затем высушивают, смачивают в растворе крахмала и снова высушивают.
Йодометрическое определение
42.3. Сульфиты окисляются раствором йода до сульфатов. Количество йода, необходимое для окисления, находят по разности между прибавленным его количеством и оставшимся, определяемым титрованием тиосульфатом. При отсутствии мешающих влияний сульфиты определяются прямо (вариант А), в противном случае двуокись серы удаляют подкислением и переводят током азота в поглощающий раствор едкого натра с глицерином, где ее определяют йодометрически (вариант Б).
В зависимости от содержания сульфитов в пробе подбирают возможные для определения концентрации титрованных растворов и количество пробы, отбираемой для анализа. Определение можно практически проводить при концентрации сульфитов в 1 л, равной 0,5 мг и выше, если обрабатывается 200 - 300 мл пробы. При применении 0,02 н. растворов точность определения равна приблизительно +/- 0,2 мг в 1 л воды.
Мешающие влияния
42.4. Определению по варианту А мешают органические вещества, взаимодействующие с йодом, мешают и сероводород, нитриты, железо. В их присутствии определение проводится по варианту Б.
Определению по варианту Б мешает сероводород. Сероводород удаляют в виде H2S прибавлением 1 мл насыщенного раствора хлорида двухвалентной ртути (Осторожно, яд!) к пробе перед ее подкислением или при ее отборе.
Аппаратура
42.5. Склянка с притертой пробкой емкостью 200 - 300 мл (калибрование см. стр. 76).
Колба емкостью 1 л, снабженная капельной воронкой; подводящая газ трубка доходит до дна колбы; отводная трубка соединена с промывалками газа (для определения по варианту Б).
Промывалки газа емкостью 250 мл со стеклянными фильтрующими пластинками (при определении по варианту Б).
Стальной баллон с азотом (при определении по варианту Б).
Реактивы
42.6. Фосфорная кислота, 25%-ный раствор: 175 мл 85%-ной H3PO4 ч.д.а. смешивают с дистиллированной водой и доводят до 1 л.
Йод, приблизительно 0,1 н. раствор: 12,7 г очищенного возгонкой йода растворяют совместно с 25 г KJ в 30 - 40 мл дистиллированной воды и доводят до 1 л.
Йод, приблизительно 0,02 н. раствор: 200 мл 0,1 н. раствора йода разбавляют до 1 л дистиллированной водой.
Тиосульфат натрия, 0,1 н. раствор: 24,8 г Na2S2O3·5H2O ч.д.а. растворяют в 900 мл дистиллированной воды, прибавляют 0,2 г Na2CO3 ч.д.а. и после растворения доводят дистиллированной водой до 1 л. Поправку к титру определяют способом, приведенным при определении кислорода, по 0,1 н. титрованному раствору бихромата (4,9037 г K2Cr2O7 ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и доводят при 20 °C до 1 л).
Тиосульфат натрия, 0,02 н. раствор: 200 мл 0,1 н. раствора тиосульфата смешивают с 800 мл дистиллированной воды, в которой предварительно растворяют 0,2 г Na2CO3 ч.д.а. Поправку к титру раствора определяют способом, приведенным при определении кислорода по 0,02 н. основному раствору бихромата (0,9807 г K2Cr2O7 ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и доводят при 20 °C до 1 л).
Крахмал, 0,5%-ный раствор (приготовление см. стр. 63).
Едкий натр, 0,1 н. раствор в 5%-ном глицерине: 4 г NaOH ч.д.а. растворяют в дистиллированной воде, прибавляют 50 мл глицерина и доводят до 1 л (для определения по варианту Б).
Ход определения
42.7. Вариант А (прямое титрование пробы). Склянку известного объема наполняют до горлышка пробой. Пипеткой вводят на дно склянки 3,0 - 10,0 мл раствора йода (0,1 н. или 0,02 н.). Другой пипеткой к пробе прибавляют 3 мл фосфорной кислоты. Склянку закрывают так, чтобы под пробкой не оставалось пузырьков воздуха, содержимое склянки перемешивают переворачиванием и через 5 мин стояния в темноте количественно переносят в колбу для титрования. Титруют 0,1 н. или 0,02 н. раствором тиосульфата до светло-желтой окраски. Затем прибавляют раствор крахмала и титруют до обесцвечивания.
Консервированную пробу переносят количественно в колбу для титрования с притертой пробкой, прибавляют 3,0 - 10,0 мл 0,1 н. или 0,02 н. раствора йода и 5 мл фосфорной кислоты. Титруют через 5 мин стояния в темноте способом, описанным выше.
Одновременно находят объем раствора тиосульфата, расходуемого при холостом определении с дистиллированной водой, которое проводят с тем же объемом раствора йода, что и при анализе пробы.
Вариант Б (определение после перегонки SO2 в раствор щелочи). В две промывалки отмеривают по 50 мл 0,1 н. раствора едкого натра с глицерином, присоединяют одну к другой и соединяют с отводной трубкой колбы, в которую предварительно наливают консервированную пробу (200 - 300 мл). Из колбы и промывалок воздух удаляют слабым током азота, который пропускают приблизительно 2 мин. Потом на каждые 100 мл пробы прибавляют через капельную воронку по 10 мл разбавленной фосфорной кислоты. Смесь нагревают в течение 20 - 30 мин при 60 - 80 °C, продолжая непрерывно пропускать слабую струю азота. Затем промывалки отъединяют, а их содержимое количественно переносят в колбу для титрования, снабженную притертой пробкой. К раствору прибавляют 3 - 10 мл 0,1 н. или 0,02 н. раствора йода, подкисляют 10 мл разбавленной фосфорной кислоты и через 5 мин стояния в темноте титруют 0,1 н. или 0,02 н. титрованным раствором тиосульфата с индикатором крахмалом.
Одновременно определяют расход тиосульфата на холостой опыт: к 100 мл 0,1 н. раствора едкого натра с глицерином прибавляют 10 мл разбавленной фосфорной кислоты и такой же объем раствора йода, как при анализе пробы, и титруют раствором тиосульфата той же нормальности.
Расчет. Содержание сульфит-ионов (x) в мг/л, определенное по обоим вариантам, вычисляют по формуле

где a - расход раствора тиосульфата при холостом определении, мл;
b - расход раствора тиосульфата при титровании пробы, мл;
k - поправка для приведения концентрации раствора тиосульфата к точно 0,1 н. или 0,02 н.;
N - нормальность раствора тиосульфата;
V1 - объем склянки для пробы, мл;
V2 - объем раствора йода, прибавленного к пробе, мл (только по варианту А);
V3 - объем фосфорной кислоты, прибавленной к пробе, мл (только по варианту А).
Если анализировалась консервированная проба, то:
V2 - объем прибавленного раствора едкого натра, мл;
V3 - объем прибавленного глицерина, мл.
Округление результатов
Диапазон, мг/л | 0,5 - 5,0 | 5,0 - 10,0 | 10,0 - 20,0 | 20,0 - 50,0 |
Округление, мг/л | 0,1 | 0,2 | 0,5 | 1 |
Колориметрическое определение с фуксином
42.8. Двуокись серы, выделенная из пробы подкислением фосфорной кислотой, переводится при 60 - 80 °C струей азота в поглотительный раствор едкого натра с глицерином. Образующийся сульфит дает в кислой среде с фуксин-формальдегидным реактивом красно-фиолетовое окрашивание, интенсивность которого прямо пропорциональна концентрации сульфита. Чувствительность определения - 0,0002 мг сульфита в 1 мл поглотительного раствора. Если взять для анализа 25 мл этого раствора и 250 мл пробы, можно определить сульфиты в концентрации 0,02 мг в 1 л воды и выше.
Мешающие влияния
42.9. Определению мешает сероводород. Влияние его устраняют прибавлением к пробе 1 мл насыщенного раствора хлорида ртути (II) перед подкислением пробы.
Аппаратура
42.10. Склянки с притертыми пробками емкостью 200 - 300 мл (калибрование см. стр. 76).
Колба с капельной воронкой (см. стр. 208).
Поглотители со стеклянными фильтрующими пластинками емкостью 30 мл.
Фотометр, светофильтр  .
.
.Кюветы толщиной 3 - 5 см.
Реактивы
42.11. Едкий натр, 0,1 н. раствор с 5% глицерина (приготовление см. стр. 208).
Фосфорная кислота, 25%-ный раствор (приготовление см. стр. 208).
Фуксин: а) запасной раствор: 0,65 г основного фуксина ч.д.а. растворяют в 20 мл этилового спирта и разбавляют приблизительно до 100 мл дистиллированной водой. Медленно прибавляют 60 мл концентрированной H2SO4 ч.д.а., а после охлаждения дополняют дистиллированной водой до 1 л. Раствор два раза встряхивают с бензином, прибавляя его порциями по 20 мл. После отделения бензинового слоя водный раствор встряхивают с 2 - 3 г активного угля и фильтруют через плотный фильтр с добавлением 5 г волокнистого асбеста. Затем раствор переносят в кювету толщиной 5 см и измеряют оптическую плотность при 600 - 750 нм. Если величина оптической плотности превысит 0,02, раствор снова фильтруют через тот же фильтр, пока оптическая плотность не понизится до указанной величины.
Формальдегид ч.д.а., 2%-ный раствор.
Фуксин-формальдегидный реактив: 10 частей раствора фуксина смешивают с 1 частью 2%-ного раствора формальдегида и тщательно перемешивают. Смесь приготовляют не раньше чем за 10 мин до применения.
Сульфит натрия, стандартный раствор:
а) запасной: 0,20 г безводного Na2SO3 ч.д.а. растворяют в 1 л 0,1 н. раствора едкого натра, содержащего 5% глицерина. Раствор сохраняется в течение 1 суток; его пригодность проверяют йодометрически. На титрование 100 мл этого раствора теоретически должно расходоваться 12,49 мл 0,02 н. раствора йода; в соответствии с результатом этого титрования раствор затем разбавляют так, чтобы 1 мл содержал 0,10 мл  ;
;
б) рабочий: 10 мл запасного раствора разбавляют до 100 мл 0,1 н. раствором едкого натра, содержащим 5% глицерина; раствор сохраняется в течение 1 суток, 1 мл его содержит 0,010 мг  .
.
Калибровочная кривая
42.12. В мерные колбы емкостью 25 мл отмеряют 0 - 0,5 - 1,0 - 1,5 - 2,0 - 2,5 мл рабочего раствора сульфита и дополняют 0,1 н. раствором едкого натра до метки. Растворы, отвечающие концентрациям 0,0002 - 0,001 мг сульфитов в 1 мл, применяют при использовании кювет толщиной 5 см. Если применяют кюветы с меньшим расстоянием между стенками, то приготовляют более концентрированные растворы.
В несколько колб наливают по 15 мл приготовленных стандартных растворов, прибавляют по 10 мл свежеприготовленного реактива и через 30 мин измеряют оптическую плотность, вычитают значения ее для холостого определения и строят график зависимости оптической плотности от концентрации сульфита.
Ход определения
42.13. В колбу помещают консервированную пробу. В два поглотителя наливают по 5 мл 0,1 н. раствора едкого натра, содержащего 5% глицерина. Поглотители соединяют один с другим и подсоединяют к отводной газовой трубке колбы. Затем из колбы удаляют воздух пропусканием слабой струи азота. Пропускание азота ведут около 2 мин, потом через капельную воронку прибавляют по 10 мл фосфорной кислоты на каждые 100 мл пробы. Смесь нагревают 20 - 30 мин при 60 - 80 °C, непрерывно пропуская струю азота. Затем поглотители отсоединяют, их содержимое количественно смывают в две мерные колбы емкостью 25 мл, доливают 0,1 н. раствором едкого натра до метки и тщательно перемешивают.
В одну колбу емкостью 50 мл наливают 15 мл 0,1 н. раствора едкого натра, в другие две такие же колбы - по 15 мл раствора из мерных колб. Затем прибавляют по 10 мл свежеприготовленного фуксин-формальдегидного реактива и через 30 мин измеряют оптические плотности растворов, вводят поправку на холостое определение и по калибровочной кривой находят содержание сульфит-ионов.
Расчет. Содержание сульфит-ионов (x) в мг/л вычисляют по формуле

где c1, c2 - концентрации сульфитов в первом и втором поглотительных растворах, найденные по калибровочной кривой, мл;
V - объем пробы, взятой для анализа (после вычитания объема консервирующих реактивов), мл;
25 - емкость мерной колбы, мл.
Округление результатов
Диапазон, мг/л | 0,020 - 0,05 | 0,05 - 0,10 | 0,10 - 0,50 | 0,50 - 1,00 |
Округление, мг/л | 0,005 | 0,01 | 0,05 | 0,1 |
Общие положения
43.1. Фенолы являются ароматическими соединениями с одной или несколькими гидроксильными группами в бензольном кольце.
Фенол, крезолы, ксилолы, гваякол, тимол и некоторые другие соединения этой группы можно отделить от остальных фенолов и других мешающих веществ, присутствующих в пробе, отгонкой водяным паром при определенных условиях. Летучие фенолы являются основным компонентом фенольных сточных вод, и вместе с ними они попадают в канализационную сеть и в поверхностные воды, загрязняя их. В концентрациях, равных нескольким миллиграммам на 1 л, они могут влиять на биологическую жизнь рек. Некоторые из фенолов в концентрациях порядка нескольких тысячных долей миллиграмма на 1 л являются причиной неприятного хлорфенольного запаха и привкуса, появляющихся в результате хлорирования поверхностных вод в процессе водоподготовки.
Для определения летучих фенолов (обычно смеси неопределенного состава) рекомендуется колориметрический метод с амидопирином (пирамидоном).
Пробы воды, содержащие более 100 мг/л фенолов, не консервируют; их надо анализировать не позже чем через 5 суток после отбора. Пробы, содержащие менее 100 мг/л фенолов, если их не анализируют в день отбора, следует консервировать прибавлением 4 г едкого натра на 1 л пробы. Пробы, содержащие менее 0,05 мг/л фенола, надо обрабатывать сразу же после их отбора.
Результаты определения выражают в миллиграммах фенола на 1 л воды.
Колориметрическое определение фенолов с пирамидоном
(диметиламинантипирином)
43.2. Метод основан на образовании окрашенных соединений фенола с пирамидоном в присутствии в качестве окислителя персульфата в щелочной среде в интервале pH примерно от 9,3 до 10,0.
Метод дает возможность определить суммарное содержание фенольных соединений и их производных (в частности, продуктов хлорирования фенолов) от 0,001 мг/л фенолов и выше. Точность определения составляет +/- 10%.
Аппаратура
43.3. Перегонный аппарат, состоящий из стеклянной колбы емкостью 1 л и холодильника Либиха.
Делительные воронки емкостью 150, 500 и 1200 мл.
Стеклянные колбы емкостью 200 - 250 мл.
Фотометр с набором кювет  или цилиндры Геннера.
или цилиндры Геннера.
или цилиндры Геннера.Реактивы
43.4. Все реактивы готовят на дистиллированной воде, не содержащей фенола и свободного хлора.
Серная кислота, пл. 1,84.
Буферный раствор (pH = 9,3), 50 г хлорида аммония растворяют в 900 мл дистиллированной воды, добавляют 40,0 мл концентрированного раствора аммиака и доводят объем дистиллированной воды до 1 л.
Пирамидон, 3 - 5%-ный раствор, раствор пригоден к употреблению в течение 3 - 5 дней.
Персульфат аммония, 20%-ный раствор, растворяют 50 г персульфата аммония в 200 мл воды, нейтрализуют концентрированным раствором аммиака по лакмусовой бумаге, разбавляют до 250 мл и фильтруют. Раствор устойчив в течение 30 суток.
Экстракционная смесь. Смешивают 100 мл хлороформа с 200 мл изоамилового спирта.
Сульфат меди, 10%-ный раствор.
Фенол, стандартный раствор:
а) запасной: растворяют 10 г фенола в 1 л дистиллированной воды. В 1 мл этого раствора должно содержаться 10 мг фенола. Концентрацию раствора проверяют бромированием отобранной порции его и йодометрическим титрованием <*>;
б) рабочий I: (0,01 мг фенола в 1 мл); 1 мл основного стандартного раствора фенола разбавляют дистиллированной водой до 1 л; раствор приготовляют в день анализов (1 мл раствора содержит 0,01 мг фенола);
в) рабочий II: 50 мл рабочего раствора I разбавляют дистиллированной водой до 500 мл; раствор приготовляют непосредственно перед употреблением; 1 мл раствора содержит 0,001 мг фенола.
--------------------------------
<*> Ю.Ю. Лурье и А.И. Рыбникова. Химический анализ производственных сточных вод. М., Госхимиздат, 1966, стр. 224.
Калибровочная кривая
43.5. Калибровочную кривую строят для интервала 0 - 0,10 мг/л фенола, обрабатывая 500 мл стандартных растворов способом, описанным в ходе определения (без дистилляции). Определяют оптическую плотность, вводят поправку на холостое определение и строят график зависимости оптической плотности от концентрации.
Ход определения
43.6. Количественному определению фенолов должна предшествовать их отгонка из анализируемой пробы для выделения только летучих с водяным паром фенолов и исключения мешающего влияния других соединений.
В зависимости от ожидаемой концентрации фенолов берут следующие количества воды для анализа:
Концентрация фенола, мг/л | 0,001 - 0,005 | 0,005 - 0,05 | 0,05 - 0,10 | 0,25 - 0,50 |
Объем воды для анализа, мл | 1000 | 500 | 300 | 100 |
Для связывания сульфидных соединений добавляют 10%-ный раствор сульфата меди из расчета 1,0 мл на каждые 100 мл воды. Фенолы отгоняют из сильнокислой среды (pH = 2,0), для чего добавляют по 1 мл концентрированной серной кислоты на каждые 100 мл анализируемой воды.
В колбу-приемник наливают 10 - 15 мл 0,01 н. раствора едкого натра и отгоняют первые фракции, помещая конец холодильника в щелочной раствор. При начальных объемах 1000, 500 и 300 мл отгоняют соответственно 800, 400 и 250 мл. Если на анализ взята проба объемом 100 мл, то предварительно ее разбавляют дистиллированной водой до 250 мл и отгоняют 200 мл. Дистиллят переносят в мерную колбу соответствующей емкости, доводят до метки дистиллированной водой и отбирают аликвотную часть.
Для ориентировочного определения фенолов в делительную воронку объемом 250 мл помещают 100 мл дистиллята, добавляют 2 мл буферного раствора, тщательно перемешивают и затем прибавляют 1 мл раствора пирамидона и 3 мл раствора персульфата аммония и снова перемешивают.
Одновременно готовят шкалу стандартных растворов и проводят "холостой" опыт. Через 30 мин вносят во все делительные воронки по 8 мл экстракционной смеси и встряхивают в течение 2 мин. Примерное количество фенолов в пробе оценивают по интенсивности окраски органического слоя.
Для количественного определения все действия проводят точно так же. Количество реактивов, применяемых при определении, берут в зависимости от объема дистиллята, взятого для анализа, согласно табл. 10.
Таблица 10
в зависимости от объема дистиллята
Определение | Объем дистиллята, мл | Буферный раствор, мл | Раствор пирамидона, мл | Раствор персульфата аммония, мл | Экстракционная смесь, мл |
Без экстракции | 100 | 2 | 1 | 3 | - |
С экстракцией | 100 | 2 | 0,3 | 3 | 8 |
То же | 500 | 10 | 1,5 | 15 | 20 |
" | 1000 | 20 | 3,0 | 30 <*> | 25 |
--------------------------------
Окрашенный экстракт пропускают через бумажный фильтр (белая лента) для удаления водной эмульсии в органическом слое и повышения устойчивости окраски во времени (окраска экстракта устойчива в течение 4 ч).
Величину оптической плотности окрашенных экстрактов исследуемых образцов, "холостого" опыта и шкалы стандартных растворов определяют на ФЭК-Н-57 или ФЭК-М; можно пользоваться также цилиндрами Геннера.
При концентрации фенола 0,02 - 0,03 мг/л и выше образуется интенсивная окраска водного раствора продукта реакции фенола с пирамидоном; интенсивность водного раствора без экстракции определяют двумя методами:
1) измерением оптической плотности водных растворов с помощью фотоэлектроколориметра при длине волны 540 нм (зеленый светофильтр в кювете с толщиной слоя, равной 50 мм);
2) визуальным сравнением в цилиндрах Геннера интенсивности окраски исследуемой пробы воды с окраской соответствующих стандартных растворов.
При концентрации фенола меньшей 0,02 мг/л интенсивность окраски определяют только после экстракции окрашенного вещества смесью хлороформа с изоамиловым спиртом. Величину оптической плотности органических экстрактов измеряют при светофильтре  в кювете с толщиной слоя 10 мм.
в кювете с толщиной слоя 10 мм.
в кювете с толщиной слоя 10 мм.Концентрацию фенола находят по калибровочной кривой.
Расчет. Содержание летучих фенолов (x) в мг/л вычисляют по формуле

где c - концентрация фенола, найденная по калибровочной кривой, мг/л;
V1 - объем пробы, взятой для определения, мл;
V2 - общий объем дистиллята, мл;
V3 - объем дистиллята, взятый для экстракции, мл.
Общие положения
44.1. Содержание соединений фосфора в воде невелико и колеблется в довольно узких пределах. В грунтовых водах растворенные соли ортофосфорной кислоты, обычно определяемые анализом, содержатся в концентрациях порядка 0,001 - 0,1 мг/л. В поверхностных загрязненных водах количество фосфатов может возрастать до десятых долей миллиграмма, в исключительных случаях до нескольких миллиграммов в 1 л.
В поверхностные воды фосфаты попадают главным образом из почвы при разложении органических веществ, а также со сбросом малоочищенных и неочищенных сточных вод, содержащих фосфорные соединения.
При некоторых способах обработки воды, а также со сточными водами, содержащими синтетические моющие средства, в поверхностные воды могут попадать полифосфаты, которые в естественных условиях медленно гидролизуются до ортофосфатов. Выражение "общий фосфор" охватывает все виды фосфатов, содержащихся в воде, а именно: растворенные и нерастворенные, неорганические и органические связанные фосфаты.
Фосфаты рекомендуется определять по возможности непосредственно после отбора пробы.
Для определения растворенных ортофосфатов и полифосфатов пробу необходимо фильтровать через мембранный фильтр N 3 или плотный бумажный фильтр с синей лентой на месте отбора пробы. Для приостановления биохимических процессов пробу можно консервировать, добавляя 2 - 4 мг хлороформа на 1 л воды.
Результаты определения фосфатов и полифосфатов выражают в миллиграммах  на 1 л или в миллиграмм-эквивалентах соответствующей ионной формы на 1 л воды. Результаты определения "общего" фосфора выражают в миллиграммах
на 1 л или в миллиграмм-эквивалентах соответствующей ионной формы на 1 л воды. Результаты определения "общего" фосфора выражают в миллиграммах  в 1 л воды.
в 1 л воды.
Колориметрическое определение растворенных ортофосфатов
44.2. Желтое комплексное соединение, образующееся при взаимодействии ортофосфат-ионов с молибдатом в кислой среде, под действием восстановителей превращается в интенсивно окрашенное синее соединение. Из всех применяемых для этой цели восстановителей наиболее устойчивые окраски дает аскорбиновая кислота.
Однако восстановление аскорбиновой кислотой происходит только при повышенной температуре, т.е. в условиях, когда полифосфаты и органические эфиры фосфорной кислоты гидролизуются с образованием ортофосфорной кислоты.
Введение в раствор соли сурьмы приводит к образованию более сложного соединения, в состав которого входит сурьма в отношении Sb:P = 1:1, и реакция тогда происходит быстро при комнатной температуре. Повышается и интенсивность окраски.
Полифосфаты и сложные эфиры фосфорной кислоты в этих условиях в реакцию не вступают.
При проведении определения в кюветах с толщиной слоя 5 см можно без разбавления пробы определять не более 1,00 мг/л  . Точность определения в диапазоне концентрацией от 0,02 до 1,00 мг/л
. Точность определения в диапазоне концентрацией от 0,02 до 1,00 мг/л  составляет +/- 0,01 - 0,02 мг/л; чувствительность - 0,01 мг/л
составляет +/- 0,01 - 0,02 мг/л; чувствительность - 0,01 мг/л 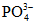 .
.
Мешающие влияния
44.3. Этим способом можно определять ортофосфат-ионы в присутствии многих неорганических и органических веществ, в том числе и в присутствии полифосфатов и сложных эфиров фосфорной кислоты. Изменение некоторых внешних условий (температуры в пределах +/- 10 °C и освещения) не оказывает влияния на результат определения.
Можно изменять и концентрации добавляемых реактивов (понизить до 50% или повысить до 100%); следует только учитывать изменение объема жидкости. Очень важной особенностью метода является то, что реактивы можно прибавлять и в обратном порядке.
Сильнокислые и сильнощелочные пробы предварительно нейтрализуют.
Определению мешают сульфиды и сероводород в концентрациях, превышающих 3 мг/л S2-; мешающее влияние можно устранить прибавлением нескольких миллиграммов твердого KMnO4 на 100 мл пробы и встряхиванием в течение 1 - 2 мин; раствор должен остаться розовым. После этого прибавление реактивов надо проводить в обратном порядке; сначала прилить раствор аскорбиновой кислоты, перемешать, затем прибавить раствор молибдата.
Определению мешают также хроматы в концентрациях, превышающих 2 мг/л 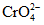 . Это мешающее влияние устраняют, приливая реактивы в обратном порядке, как и в предыдущем случае.
. Это мешающее влияние устраняют, приливая реактивы в обратном порядке, как и в предыдущем случае.
Мешают определению арсенаты. Обычно в водах арсенаты отсутствуют, но если они окажутся в больших количествах, надо, определив их содержание, вычесть результат из найденного содержания фосфатов.
Мешающее влияние нитритов устраняют сульфаминовой кислотой, которую вводят в состав применяемого реактива. При большом содержании железа следует ввести эквивалентное количество комплексона III.
Аппаратура
44.4. Фотометр со светофильтром  .
.
.Кюветы толщиной слоя 2 - 4 см.
Реактивы
44.5. Смесь молибдата аммония, серной кислоты, сульфаминовой кислоты, хлорида сурьмы и винной кислоты. К 300 мл бидистиллята приливают при перемешивании 144 мл концентрированной серной кислоты ч.д.а. Затем охлаждают приблизительно до 20 °C и при перемешивании прибавляют 10 г сульфаминовой кислоты (H2NSO3H) ч.д.а., растворенной в 100 мл дистиллированной воды, раствор 0,235 г хлорида сурьмы SbCl3 ч.д.а. и 0,6 г винной кислоты ч.д.а. (или 0,345 г антимонилтартрата калия KSbOC4H4·0,5H2O ч.д.а.) в 100 мл дистиллированной воды, после чего разбавляют полученную смесь до 1 л дистиллированной водой. Реактив хранят в склянке из оранжевого стекла.
Аскорбиновая кислота, 10%-ный раствор. Растворяют 10 г возможно более чистой аскорбиновой кислоты в бидистилляте и разбавляют раствор до 100 мл. Хранят раствор на холоде; он устойчив приблизительно 30 дней.
Фосфат калия однозамещенный, стандартный раствор:
а) запасной раствор: растворяют 0,7165 г KH2PO4 ч.д.а., высушенного в течение 2 ч при 105 °C, в дистиллированной воде, прибавляют 2 мл хлороформа и разбавляют дистиллированной водой до 1 л; 1 мл этого раствора содержит 0,50 мг  ;
;
б) рабочий раствор I: разбавляют 10 мл запасного раствора до 1 л дистиллированной водой; применяют всегда свежеприготовленный раствор; 1 мл его содержит 0,05 мг  ;
;
в) рабочий раствор II: разбавляют 50 мл рабочего раствора I до 250 мл дистиллированной водой; применяют всегда свежеприготовленный раствор; 1 мл его содержит 0,01 мг 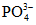 .
.
Калибровочная кривая
44.6. В ряд колб помещают 0; 1,0; 2,5; 5,0; 10,0; 25,0; 50 мл рабочего стандартного раствора II и объем всех растворов доводят до 50 мл дистиллированной водой. В полученных растворах, концентрации которых отвечают 0; 0,02; 0,05; 0,10; 0,20; 0,50; 1,00 мг/л  , определяют фосфаты описанным ниже способом. Вводят поправку на "холостой опыт" и строят калибровочную кривую в координатах оптическая плотность - концентрация фосфат-ионов.
, определяют фосфаты описанным ниже способом. Вводят поправку на "холостой опыт" и строят калибровочную кривую в координатах оптическая плотность - концентрация фосфат-ионов.
Ход определения
44.7. К 50 мл пробы, профильтрованной в день отбора (на месте отбора или в лаборатории) через плотный фильтр (синяя лента), или к меньшему объему пробы, но разбавленному до 50 мл дистиллированной водой, приливают сначала 2 мл раствора молибдата, через короткое время 0,5 мл раствора аскорбиновой кислоты. Смесь перемешивают. Одновременно проводят "холостое" определение с 50 мл дистиллированной воды. Если анализируемая проба содержит полифосфаты и органические соединения фосфора, измеряют оптическую плотность в промежутке времени от 5 до 15 мин после добавления раствора аскорбиновой кислоты.
Если легко гидролизующихся полифосфатов нет, этот промежуток времени может быть увеличен до 60 мин.
Если же нет ни полифосфатов, ни органических фосфатов, измерение оптической плотности можно проводить в течение времени от 5 мин до 48 ч после добавления аскорбиновой кислоты.
Из найденной величины оптической плотности вычитают оптическую плотность "холостого" определения. Если проба была несколько мутной или окрашенной, надо также вычесть оптическую плотность раствора, получаемого после добавления молибдата, но перед введением аскорбиновой кислоты. Содержание фосфатов находят по калибровочной кривой.
Расчет. Содержание растворенных неорганических ортофосфатов  (x) в мг/л вычисляют по формуле
(x) в мг/л вычисляют по формуле

где c - концентрация фосфат-ионов, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мл;
50 - объем, до которого разбавляют раствор, мл.
Округление результатов
Диапазон, мг/л | 0,01 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 | 1,0 - 2,0 |
Округление, мг/л | 0,01 | 0,02 | 0,05 | 0,1 |
Определение гидролизующихся полифосфатов
44.8. В методе используется кислый гидролиз полифосфатов, при котором они переходят в растворимые неорганические ортофосфаты, определяемые с помощью молибдата и аскорбиновой кислоты. Одновременно определяют ортофосфаты, первоначально бывшие в пробе, и содержание их вычитают из найденного результата.
Мешающие влияния
44.9. Мешают те же причины, что и в случае определения растворенных неорганических ортофосфатов. При гидролизе возможно также частичное разрушение органических соединений фосфора, особенно при высоком содержании этих веществ.
Аппаратура
44.10. Чашки, стаканы или колбы емкостью 200 мл из химически стойкого стекла, желательно кварцевого, не содержащего фосфор.
Фотометр такой же, как и для определения неорганических ортофосфатов (см. стр. 217).
Реактивы
44.11. Те же, что и для колориметрического определения неорганических ортофосфатов.
Ход определения
44.12. К 100 мл пробы, профильтрованной через плотный фильтр (синяя лента) на месте отбора или в тот же день в лаборатории, или к меньшему объему, разбавленному до 100 мл дистиллированной водой, добавляют 2,0 мл 37%-ного раствора серной кислоты и кипятят 30 мин. Объем пробы поддерживают постоянным, добавляя дистиллированную воду в пределах 50 - 90 мл.
После охлаждения до комнатной температуры переводят раствор в мерную колбу емкостью 100 мл и разбавляют дистиллированной водой до метки.
Отбирают пипеткой 50 мл и определяют ортофосфаты, как описано выше (см. стр. 218).
Расчет. Содержание гидролизующихся полифосфатов (x) в мг/л вычисляют по формуле
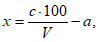
где c - концентрация  после гидролиза, найденная по калибровочной кривой, мг/л;
после гидролиза, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для определения, мг;
a - концентрация неорганических ортофосфатов, найденная в пробе отдельным определением, мг/л;
100 - объем, до которого разбавлена проба, мл.
Округление результатов
Диапазон, мг/л | 0,10 - 0,20 | 0,20 - 0,50 | 0,50 - 1,0 |
Округление, мг/л | 0,02 | 0,05 | 0,1 |
Колориметрическое определение общего содержания фосфора
("общего фосфора")
44.13. Мокрым сжиганием в присутствии серной кислоты и перекиси водорода все виды фосфатов в пробе переводятся в растворимые неорганические ортофосфаты, которые определяют затем молибдатом с аскорбиновой кислотой.
Мешающие влияния
44.14. Мешающие влияния те же, что и при определении ортофосфатов. Нитриты и таннин не мешают определению. Большое количество органических веществ мешает полному сжиганию. В этом случае надо пробу сначала разбавить или применить весовое определение.
Органические соединения фосфора, в молекулах которых атомы фосфора непосредственно связаны с атомами углерода, описываемым способом минерализации, не разлагаются.
Аппаратура
44.15. Чашки, стаканы или колбы емкостью приблизительно 100 мл из стойкого стекла, желательно кварцевого, не содержащего фосфора.
Фотометр и кюветы такие же, как и при определении ортофосфатов (см. стр. 217).
Сушильный шкаф (160 °C).
Реактивы
44.16. Серная кислота, 37%-ный раствор: растворяют 337 мл 98%-ной H2SO4 ч.д.а. при перемешивании и охлаждении в 600 мл дистиллированной воды и доводят до 1 л.
Перекись водорода, 30%-ный раствор, не стабилизированный добавлением фосфатов.
Остальные реактивы те же, что и при определении растворенных ортофосфатов (стр. 217).
Калибровочная кривая
Та же, что и для определения растворенных ортофосфатов (см. стр. 218).
Ход определения
44.17. Отмеривают 50 мл нефильтрованной, хорошо перемешанной пробы (или меньшее количество ее, доведенное до 50 мл дистиллированной водой) в чашку, стакан или колбу из химически стойкого стекла. Добавляют 0,15 мл (3 капли) перекиси водорода и точно 1,0 мл 37%-ного раствора серной кислоты. Смесь нагревают и выпаривают при температуре 160 °C в течение 6 ч. Минерализованная проба должна получиться чистой и бесцветной, в противном случае надо повторить минерализацию, взяв для анализа более разбавленный раствор.
После охлаждения содержимое чашек доводят до 30 мл дистиллированной водой, кипятят 2 - 3 мин, охлаждают и количественно переводят в колбу, доводя объем в колбе до 50 мл.
Затем определяют содержание ортофосфат-ионов, как описано на стр. 218.
Расчет. Содержание "общего фосфора" (x) в мг/л вычисляют по формуле

где c - концентрация  , найденная по калибровочной кривой, мг/л;
, найденная по калибровочной кривой, мг/л;
, найденная по калибровочной кривой, мг/л;V - объем пробы, взятой для определения, мл;
50 - объем, до которого разбавлена проба, мл.
Округление результатов. Результаты округляют так же, как и при определении растворенных неорганических ортофосфатов.
Общие положения
45.1. В поверхностных водах обычно встречаются небольшие количества фторидов; в подземных водах концентрация их доходит до 10 мг/л и выше в зависимости от геологических условий. Повышенным содержанием фторидов отличаются сточные воды предприятий стекольной и химической промышленности, некоторые виды шахтных вод, воды от рудообогатительных фабрик и др.
Присутствие фторидов в питьевой воде имеет в первую очередь гигиеническое значение. При употреблении воды с содержанием фторидов, превышающим 1,5 мг/л, возникает заболевание зубов - флюороз, или "крапчатость" эмали; при полном отсутствии в питьевой воде фторидов развивается кариез зубов.
Для определения фторидов в питьевых водах, поверхностных и подземных источниках приведен цирконий-ализариновый колориметрический метод в двух вариантах:
а) визуальным сравнением в цилиндрах Несслера из бесцветного стекла емкостью 100 мл, при концентрации фтора в воде 0,05 до 1,4 мг/л;
б) с помощью фотометра для концентрации фтора от 0,05 до 1,5 мг/л.
Для определения фтора в окрашенных и мутных водах или в присутствии веществ, мешающих определению, приводится метод колориметрического определения с перегонкой.
Пробы воды отбирают в бутыли из специального стекла или полиэтилена. Пробы не консервируют. Результаты определения приводят в миллиграммах фторидов на 1 л воды.
Цирконий-ализариновый метод
45.2. При реакции фторидов с цирконием (IV) образуются комплексные фтористые соли. Устойчивость таких комплексов больше, чем устойчивость цветных комплексов циркония (IV) с органическими красителями, в данном случае с ализаринсульфонатом. Выделение эквивалентного количества красителя из комплексной соли обнаруживается по изменению окраски, цвет и интенсивность которой отвечает концентрации фтора. В зависимости от концентрации фторид-иона цвет раствора изменяется от слабо-розового до желтого.
Мешающие влияния
45.3. Определению препятствует присутствие в воде хлора, который можно удалить добавлением 0,05 мл 0,5%-ного раствора арсенита натрия на каждые 0,1 мг хлора. Окрашенную и мутную воду, а также воду с повышенным содержанием органических веществ предварительно перегоняют, фториды отгоняют в виде фтористоводородной кислоты.
Некоторые вещества реагируют с реактивом или же связывают фторид-ионы, образуя комплексные соли. В присутствии 1800 - 2000 мг/л хлоридов или 0,2 мг/л алюминия возникает ошибка, равная 0,1 мг/л при 1 мг/л фторид-иона.
Наличие в воде сульфатов 300 - 400 мг/л, фосфатов более 5 мг/л или гексаметафосфата натрия 1 мг/л вызывает ошибку в определении +0,1 мг/л.
Присутствие железа (III) более 5 мг/л вызывает ошибку в определении -0,1 мг/л.
Мешающее влияние оказывает также и щелочность воды, превышающая 3 мг-экв/л.
Щелочность можно устранить прибавлением эквивалентного количества соляной или азотной кислоты.
Однако пользоваться приведенными данными для внесения поправок в полученные результаты не рекомендуется. Мешающее влияние соответствующего вещества устраняют добавлением его к ряду стандартных растворов при приготовлении растворов для калибровочной кривой. Для более точного определения фторид-иона и при наличии высоких концентраций мешающих веществ необходимо исследуемые пробы воды предварительно перегонять.
Аппаратура
45.4. Фотометр светофильтром  .
.
.Кюветы толщиной слоя воды 5 см или набор цилиндров Несслера из бесцветного стекла емкостью 100 мл.
Реактивы
45.5. Цирконий-ализариновый реактив, кислый раствор (для определения по варианту А) 0,30 г ZrOCl2·8H2O растворяют в 50 мл дистиллированной воды. В других 50 мл дистиллированной воды растворяют 0,07 г ализаринсульфоната натрия (ализаринового красного С), растворы перемешивают. Разбавленный раствор приготовляют следующим образом: 100 мл концентрированной соляной кислоты ч.д.а. смешивают с 300 мл дистиллированной воды. К другим 400 мл дистиллированной воды прибавляют 33,3 мл концентрированной H2SO4 ч.д.а. После охлаждения оба раствора кислот соединяют и перемешивают. Смесь кислот добавляют к полученному ранее раствору ализаринсульфоната и соли циркония и доводят объем до 1 л дистиллированной водой. Раствор сохраняет устойчивость в течение 6 месяцев. Раствор следует хранить в темной бутыли.
Ализариновый красный С, раствор (для определения по варианту Б): 0,75 г ализаринового красного С растворяют в дистиллированной воде и доводят объем до 1 л.
Хлорид цирконила, кислый раствор (для варианта Б): 0,354 г ZrOCl2·8H2O растворяют в 600 - 800 мл дистиллированной воды, добавляют 33,3 мл концентрированной H2SO4 ч.д.а., перемешивают и после добавления 100 мл концентрированной HCl ч.д.а. смесь снова перемешивают. После охлаждения доводят дистиллированной водой до объема 1 л. Раствором можно пользоваться не ранее чем через 1 ч после его приготовления; хранят раствор в темной бутыли.
Фторид натрия, стандартный раствор:
а) запасной: 0,221 г NaF ч.д.а., высушенного при 105 °C, растворяют в дистиллированной воде и доводят объем до 1 л; 1 мл раствора содержит 0,1 мг F-;
б) рабочий: 50 мл запасного раствора разбавляют дистиллированной водой до 1 л. Во всех случаях пользоваться свежеприготовленным раствором. I мл рабочего раствора содержит 0,005 мг F-.
Калибровочная кривая
45.6. В ряд колб наливают 0 - 1,0 - 3,0 - 5,0 - 10,0 - 15,0 - 20,0 - 25,0 - 30,0 мл рабочего раствора фторида натрия и доводят объем жидкости в каждой колбе до 100 мл дистиллированной водой. Приготовленные растворы соответствуют концентрациям 0 - 0,05 - 0,15 - 0,25 - 0,5 - 0,75 - 1,0 - 1,25 - 1,50 мг/л F-. В каждую колбу добавляют по 5 мл раствора ализаринового красного С и хлорида цирконила, перемешивают и через 1 ч колориметрируют, отмечая показания на барабане для каждой пробы.
Так же как и при анализе воды, готовят пробу дистиллированной воды для установления стрелки гальванометра на нуль.
Нулевое положение стрелки гальванометра следует проверять перед каждым определением.
Наносят на график по оси абсцисс концентрацию фторид-иона в мг/л, а по оси ординат оптическую плотность (показания барабана), соединяя полученные точки, строят калибровочную кривую. Калибровочную кривую нужно составлять всегда заново после каждого приготовления новых растворов ализарина и соли циркония.
Ход определения
45.7. Вариант А. Отмеряют в цилиндры из бесцветного стекла 0 - 1,0 - 2,0 - 3,0- ... -30,0 мл рабочего раствора фторида натрия. После доведения объема до 100 мл дистиллированной водой содержание фторид-иона в них будет 0 - 0,05 - 0,01 - 0,15- ... -1,5 мг/л. Еще в один цилиндр наливают 100 мл прозрачной пробы и после выравнивания температуры вносят в пробу и в ряд стандартных растворов по 5 мл кислого цирконий-ализаринового реактива. Смесь тщательно перемешивают и по истечении 1 ч сравнивают полученную окраску со стандартными растворами. Для каждого цикла определений необходимо приготовлять свежие стандартные растворы.
Для анализа проб с ориентировочно известной концентрацией фтора следует готовить стандартные растворы, близкие по своему значению к предполагаемой концентрации F-.
Вариант Б. Отбирают 100 мл анализируемой воды, и если она имеет сильно щелочную реакцию (pH более 8,0), то ее нейтрализуют 0,1 н. раствором HCl до перехода окраски по метиловому оранжевому. Количество 0,1 н. HCl, необходимое для нейтрализации, определяют в отдельной пробе. Затем вносят 5 мл раствора ализаринового красного С и 5 мл раствора хлорида цирконила. Тщательно перемешав, оставляют стоять в течение 1 ч в темном и прохладном месте.
Если фторид-ионов в воде содержится более 1,5 мг/л, пробу необходимо разбавить дистиллированной водой в 2 раза и более, в зависимости от концентрации их в воде.
Одновременно с анализируемой водой берут 100 мл дистиллированной воды, добавляют в нее по 5 мл растворов ализаринового красного С и хлорида цирконила, перемешивают и оставляют стоять на 1 ч вместе с пробами анализируемой воды.
Через 1 ч определяют содержание фторид-ионов, пользуясь фотометром. Для этого пробу с дистиллированной водой наливают в две кюветы, помещают их в правое и левое гнездо электрофотоколориметра и устанавливают стрелку гальванометра на нуль. Анализируемую воду наливают в третью кювету и устанавливают ее в левом гнезде. Определяют по показанию барабана оптическую плотность и по калибровочной кривой находят содержание фторид-ионов.
Расчет. Содержание фторид-ионов (x) в мг/л вычисляют по формуле

где c - концентрация фторидов, найденная по калибровочному графику или по шкале стандартных растворов, мг/л;
V - объем пробы, взятой для анализа, мл;
100 - объем, до которого разбавлена проба, мл.
Колориметрическое определение при помощи
цирконий-ализаринового реактива с предварительной отгонкой
45.8. Фториды выделяют в виде летучей кремнефтористоводородной кислоты из среды, подкисленной серной кислотой, при температуре от 135 до 145 °C. Определение в дистилляте осуществляется колориметрическим методом с цирконий-ализариновым реактивом.
Мешающие влияния
45.9. Во время перегонки необходимо проверить, не попадает ли в дистиллят повышенное количество сульфат-ионов, которые могли бы мешать определению.
Аппаратура
45.10. Перегонный аппарат состоит из круглодонной колбы емкостью 400 - 500 мл, снабженной капельной воронкой и термометром с градуировкой до 200°, опущенными до дна колбы, и парообразователя.
Реактивы
45.11. Серная кислота, концентрированная, не содержащая фторидов. Продажную кислоту кипятят в вытяжном шкафу 1 ч.
Ферросилиций, растертый в порошок, или чистый кварцевый песок. Кварцевый песок предварительно кипятят с концентрированной серной кислотой, тщательно промывают водой и высушивают.
Сульфат серебра, насыщенный раствор. Растворяют в 100 мл дистиллированной воды 1 г сульфата серебра, затем раствор фильтруют. 1 мл полученного насыщенного раствора содержит около 7 мг сульфата серебра.
Едкий натр 0,01 н. раствор.
Фенолфталеин, 0,5%-ный раствор (приготовление см. стр. 36).
Подготовка определения
45.12. Холостой опыт. В дистилляционную колбу перегонного аппарата вносят 0,5 г ферросилиция, растертого в порошок, или кварцевого песка, затем вливают 25 мл дистиллированной воды и 25 мл серной кислоты, присоединяют колбу к прибору, включая одновременно и парообразователь (к дистиллированной воде в парообразователь прибавляют 20 мл 0,1 н. раствора едкого натра). При перегонке поддерживают температуру жидкости в колбе в пределах 125 - 135 °C и в дистилляте определяют содержание фторид-ионов по варианту Б (см. стр. 224).
Проверка герметичности прибора. В дистилляционную колбу вносят снова те же реактивы и в тех же количествах, какие вводились при проведении "холостого" опыта и, кроме того, еще 5 или 10 мл стандартного рабочего раствора фторида натрия.
Проводят отгонку, как описано выше, и определяют содержание фторидов в дистилляте но методу Б (см. стр. 224).
Полученный результат не должен отклоняться от количества введенного фторида более чем на 10%.
Ход определения
45.13. Пробы с содержанием фторидов ниже 0,4 мг/л необходимо предварительно концентрировать. Для этого в фарфоровую чашку помещают 200 мл или более исследуемой воды, вносят 2 капли фенолфталеина, добавляют по каплям раствор едкого натра до нейтральной реакции и сверх того еще несколько капель, выпаривают смесь до объема 25 - 50 мл и переносят ее в перегонную колбу.
В дистилляционную колбу вливают от 25 до 100 мл анализируемой воды (в зависимости от содержания фторидов), вносят 5 - 10 стеклянных шариков, добавляют от 0,1 до 0,2 г ферросилиция или кварцевого песка, 25 мл серной кислоты и столько насыщенного раствора сульфата серебра, сколько нужно для осаждения хлоридов.
Если в ходе определения приходится вводить значительное количество сульфата серебра, то тогда такое же количество его добавляют при проведении холостого опыта. Затем присоединяют колбу к парообразователю с кипящей водой и ведут перегонку, поддерживая температуру жидкости в колбе в пределах 125 - 135 °C и пропуская пар. Перегонку заканчивают после отгонки около 200 мл и затем доводят объем дистиллятом точно до 200 мл. Определение фторидов в дистилляте проводят по вариантам А или Б (стр. 224).
Расчет. Содержание фторид-ионов (x) в мг/л вычисляют по формуле

где c - концентрация фторидов, найденная по калибровочной кривой или сравнением со стандартными растворами, мг/л;
V1 - объем пробы, взятой для перегонки, мл;
V2 - объем дистиллята, мл;
V3 - объем дистиллята, взятого для колориметрического определения, мл.
Общие положения
46.1. Содержание так называемого "активного хлора" определяется в дезинфицированной им питьевой воде. В поверхностных водах содержание хлора определяется в местах ниже сброса вод, содержащих хлор.
Понятие "активный хлор" охватывает, кроме растворенного молекулярного хлора, и другие соединения хлора, как, например, двуокись хлора, хлорамины, органические хлорамины, гипохлориты и хлориты, т.е. вещества, определяемые йодометрическим методом или при помощи о-толидина (вариант А). Из вышеупомянутых веществ в практике хлорамины и другие соединения хлора обозначаются термином "активный связанный хлор", а молекулярный хлор и гипохлориты - как "активный свободный хлор". Если нужно отличить эти две формы, пользуются методом с о-толидином (вариант Б).
Для определения "активного хлора" приведен йодометрический метод титрования и колориметрический метод с применением о-толидина. Йодометрическое определение рекомендуется для анализа вод с содержанием от 1 мг "активного хлора" в 1 л и выше - при обработке пробы объемом 200 мл. Колориметрическим методом определения пользуются в случае наличия небольшого количества пробы с концентрацией хлора от 0,01 до 7 мг/л.
Пробы, предназначенные для определения содержания хлора, не разрешается консервировать. Определение следует проводить немедленно после отбора пробы.
Результаты определения выражают в миллиграммах Cl2 в 1 л воды.
Качественное определение
46.2. Смешивают 10 мл пробы воды с 1 мл раствора о-толидина. После перемешивания возникает желтая окраска раствора при наличии не менее 0,05 мг Cl2 в 1 л.
Испытание можно также провести следующим образом. В 200 мл пробы вносят 2 мл 2 н. H2SO4 и 0,1 мл 0,2%-ного водного раствора метилового оранжевого. Параллельно с этим проводится сравнительный опыт с дистиллированной водой. В присутствии хлора в количестве, превышающем 0,1 мг/л, окраска смеси ослабнет.
Качественное определение с метиловым оранжевым можно применить и для ориентировочного количественного определения концентрации хлора. В этом случае окраску анализируемого раствора после добавления метилового оранжевого сравнивают с окрасками приготовленных стандартных растворов хлорной воды после добавления такого же количества метилового оранжевого (приготовление хлорной воды см. стр. 229). Этим способом определяется только "свободный активный хлор". Можно также титровать пробу раствором метилового оранжевого до возникновения розового окрашивания. Титр раствора метилового оранжевого устанавливается по стандартному раствору хлорной воды.
Йодометрическое определение
46.3. Хлор выделяет йод из раствора йодида. Выделенный йод титруют раствором тиосульфата по крахмалу. В кислой среде реакции протекают количественно. Титруя 0,01 н. раствором тиосульфата, можно определить 0,05 мг/л и выше свободного хлора в пробе объемом 500 мл.
Мешающие влияния
46.4. Определению хлора в питьевых или поверхностных водах с небольшим содержанием органических веществ не препятствуют нитриты, марганец и железо, если титрование осуществляется с добавлением разбавленного раствора уксусной кислоты. В присутствии большого количества органических веществ метод не дает правильных результатов.
Реактивы
46.5. Уксусная кислота, ледяная ч.д.а.
Йодид калия ч.д.а., 5%-иый раствор. Растворяют 5 г KJ в 100 мл дистиллированной воды. Полученный раствор после подкисления его 5 мл уксусной кислоты не должен окрашиваться в желтый цвет, а после добавления раствора крахмала и титрования 0,01 н. раствором тиосульфата расход последнего не должен превышать 0,2 мл.
Тиосульфат натрия 0,1 н. раствор (приготовление см. стр. 208).
Тиосульфат натрия, 0,01 н. раствор: 100 мл 0,1 н. раствора тиосульфата разбавляют до 1 л свежепрокипяченной и охлажденной дистиллированной водой с добавлением 0,2 г Na2CO3 ч.д.а. Титр определяют так же, как 0,1 н. раствора.
Крахмал, 0,5%-ный раствор (приготовление см. стр. 63).
Ход определения
46.6. При предполагаемом содержании "активного хлора" ниже 1 мг/л отмеривают 1000 мл пробы, при содержании его от 1 до 10 мг/л - 500 мл и т.д. (расход тиосульфата на титрование не должен превышать 20 мл).
Прибавляют к пробе 5 мл уксусной кислоты и около 1 г твердого йодида калия. Титрование проводят на белом фоне 0,01 н. раствором тиосульфата до получения светло-желтой окраски. Затем добавляют около 1 мл раствора крахмала и продолжают титрование до исчезновения синего окрашивания. Нельзя титровать при прямом солнечном свете. Таким же способом находят расход реактива на холостое определение с дистиллированной водой.
Расчет. Содержание активного хлора (x) в мг/л вычисляют по формуле

где a - расход 0,01 н. раствора тиосульфата, мл;
b - расход того же раствора на холостое определение, мл;
k - поправка для приведения нормальности раствора тиосульфата к точно 0,01;
V - объем анализируемой пробы, мл;
35,45 - атомный вес хлора;
0,01 - нормальность раствора тиосульфата.
Округление результатов
Диапазон, мг/л | 0,05 - 1,00 | 1,0 - 2,0 | 2,0 - 5,0 | 5,0 - 10,0 и т.д. |
Округление, мг/л | 0,05 | 0,1 | 0,2 | 0,5 |
Колориметрическое определение с о-толидином
46.7. При реакции "активного хлора" с о-толидином в кислой среде раствор окрашивается в желтый или оранжевый цвет. При концентрациях от 0,1 до 1,5 мг Cl2 в 1 л пользуются светофильтром, имеющим максимальное светопропускание в пределах от 400 до 450 нм, а при концентрациях хлора от 0,5 до 7 мг/л - светофильтром с максимумом в области 490 нм.
Реакция хлора с о-толидином отличается большой чувствительностью, позволяя при обработке пробы объемом 100 мл непосредственно определить содержание хлора в пределах от 0,01 - 7 мг/л.
Мешающие влияния
46.8. Определению мешает присутствие железа, марганца и нитритов. При концентрации железа до 0,3 мг/л, марганца до 0,01 мг/л и нитритов до 0,1 мг/л можно пренебречь их мешающим влиянием. При более высоком содержании железа и марганца в окисленной форме необходимо при точном определении хлора установить их мешающее влияние, вводя поправку на холостой опыт. В таких случаях следует пользоваться вариантом Б. Аналогичным образом поступают при наличии нитритов в концентрациях более 0,1 мг/л. Нитриты могут содержаться в водах, в которых хлор присутствует в форме хлорамина.
На определение влияют взвешенные вещества, хлорелла, мутность и собственная окраска пробы. Это воздействие можно частично устранить центрифугированием пробы перед определением или измерением оптической плотности без добавления реактива, вычитая результат этого холостого опыта из результата определения.
Аппаратура
46.9. Фотометр, светофильтры ( и
и  ).
).
и ).Кюветы длиной 1 - 5 см.
Стеклянная посуда, применяемая при определении небольших концентраций хлора, в частности конические колбы для приготовления стандартных растворов с хлорной водой, должны быть перед употреблением погружены на 3 ч в ванну с хлорной водой и потом промыты несколько раз дистиллированной водой.
Реактивы
46.10. Соляная кислота ч.д.а., разбавленная (1:6).
о-Толидин, 0,135%-ный раствор: 1,35 г - солянокислого о-толидина ч.д.а. растворяют в 500 мл дистиллированной воды и смешивают с 500 мл разбавленной соляной кислоты, приготовленной добавлением 150 мл концентрированной HCl ч.д.а. к 350 мл дистиллированной воды.
Арсенит натрия, ч.д.а., 0,5%-ный раствор.
Хлорная вода, стандартный раствор. Хлорная вода приготовляется из раствора гипохлорита натрия и дистиллированной воды, которая не должна содержать нитритов, аммиака и показывать хлоропоглощаемость (к дистиллированной воде прибавляют раствор хлорной воды, содержащей не более 1 мг "активного хлора" в 1 л, и оставляют на ночь; потом хлорированную дистиллированную воду помещают на освещенное солнцем место, пока "свободный хлор" не перестанет определяться).
В зависимости от концентрации гипохлорита натрия запасной раствор приготовляют с таким расчетом, чтобы в 1 л его содержалось около 100 мг хлора. Определив йодометрически содержание хлора в этом растворе, соответствующим разбавлением приготовляют второй стандартный раствор, содержащий 10 мг хлора в 1 л. Точное содержание хлора в этом растворе определяют также йодометрическим методом.
Фосфатный буферный раствор: 4,572 г Na2HPO4 ч.д.а., высушенного при температуре 110 °C, и 9,32 г KH2PO4 ч.д.а., высушенного при температуре 110 °C, растворяют в дистиллированной воде, тщательно перемешивают и после фильтрования доводят объем до 1 л.
Хромат калия и бихромат калия, стандартный раствор:
концентрированный: 1,55 г K2Cr2O7 ч.д.а. и 4,65 г KCrO4 ч.д.а. растворяют в фосфатном буферном растворе и доводят до 1 л этим же буферным раствором;
разбавленный: приготовляют десятикратным разбавлением концентрированного раствора фосфатным буферным раствором.
1 мл раствора соответствует по своей окраске раствору, содержащему 1 мг Cl2.
Калибровочная кривая
46.11. В мерные колбы емкостью 1 л наливают по 500 мл стандартных растворов хлорной воды с содержанием хлора от 0,02 до 0,30, или от 0,1 до 1,5, или же от 0,5 до 7,0 мг в 1 л. Для их приготовления используют стандартный раствор хлора с содержанием 10 мг Cl2 в 1 л и дистиллированную воду с нулевой хлорпоглощаемостью. Для колориметрического определения используют 95 мл каждого стандартного раствора хлора, определение проводят, как описано ниже; в остальных 400 мл определяют хлор йодометрически. На график наносят полученные значения оптической плотности против концентрации хлора, найденных йодометрическим титрованием.
Ход определения. Вариант А. (Определение хлора
при отсутствии мешающих влияний)
46.12. В мерную колбу емкостью 100 мл отмеривают 5 мл раствора о-толидина и доводят до метки пробой. По истечении 5 мин пребывания смеси в темноте при температуре 20 °C переливают ее в кювету и измеряют оптическую плотность. Если последняя выходит за пределы калибровочной кривой, тогда необходимо определение повторить при соответствующем разбавлении пробы дистиллированной водой. При анализе мутных и окрашенных проб следует провести холостое определение, при котором вместо о-толидина добавляют к пробе 5 мл разбавленной соляной кислоты. При колориметрировании пробы раствор холостого опыта помещают во вторую кювету или же измеряют его оптическую плотность по отношению к дистиллированной воде и результат вычитают из найденного значения оптической плотности пробы и по калибровочной кривой находят содержание "активного" хлора.
Вариант Б. (Определение хлора при наличии мешающих влияний)
46.13. Устанавливают сначала содержание "активного хлора" по варианту А. Затем в другую мерную колбу емкостью 100 мл наливают 5 мл раствора арсенита, 90 мл раствора о-толидина, тщательно перемешивают содержимое колбы и по истечении 5 мин измеряют оптическую плотность. По калибровочному графику находят содержание "активного хлора". Описанное выше холостое определение дает ошибку в 5%, которую (если она влияет на полученный результат) следует прибавить.
Вариант В. (Определение "связанного" (хлораминного)
и "свободного активного хлора")
46.14. В три фотометрические кюветы "a", "b" и "c" емкостью 10 мл помещают по 0,5 мл раствора о-толидина (если используются кюветы другой емкости, то отмеряют соответственно пропорциональные объемы раствора о-толидина). В ходе определения нужно добавлять раствор арсенита натрия в том же объеме, в каком прибавлен раствор о-толидина.
В кювету "a", содержащую раствор о-толидина, добавляют отмеренный объем пробы, быстро перемешивают и сейчас же (в течение 5 сек) добавляют раствор арсенита, снова быстро перемешивают и сразу же измеряют оптическую плотность. Найденный результат "a" показывает суммарную концентрацию "свободного активного хлора" и мешающих веществ.
В кювету "b", содержащую раствор арсенита, добавляют отмеренный объем пробы, быстро перемешивают и немедленно добавляют раствор о-толидина, снова быстро перемешивают и сразу же измеряют оптическую плотность (результат "b"). Измерение повторяют точно через 5 мин (результат "b"). Найденные результаты указывают влияние мешающих веществ непосредственно ("b1") и через 5 мин ("b2").
В кювету "c", содержащую раствор о-толидина, добавляют отмеренное количество пробы, быстро перемешивают и точно через 5 мин измеряют оптическую плотность (результат "c"). Этот результат указывает суммарное количество "активного хлора" и мешающих веществ.
Все результаты пересчитывают по калибровочной кривой (см. ход определения Б).
Расчет. Варианты А и Б. Содержание "активного хлора" (x) в мг/л вычисляют по формуле

где c - концентрация хлора, найденная по калибровочной кривой, мг/л;
V - объем пробы, взятой для анализа, мл.
Вариант В.
Содержание "активного хлора" (y), "свободного активного хлора (z) и "связанного активного хлора" (m) вычисляют по формулам:
y = c - b2;
z = a - b;
m = (c - b2) - (a - b1).
Округление результатов
Диапазон, мг/л | 0,02 - 0,50 | 0,50 - 1,00 | 1,0 - 2,0 | 2,0 - 5,0 и т.д. |
Округление, мг/л | 0,02 | 0,05 | 0,1 | 0,2 |
Общие положения
47.1. Хлориды являются обычно составной частью большинства природных вод. Присутствие большого количества хлоридов геологического происхождения в поверхностных водах - явление редкое. По этой причине обнаружение большого количества хлоридов служит показателем загрязнения воды бытовыми сточными и некоторыми промышленными водами.
Определение хлоридов производится в питьевых и поверхностных водах аргентометрическим титрованием по Мору или меркурометрически с применением дифенилкарбазона в качестве индикатора.
Результаты выражают в миллиграмм-эквивалентах или миллиграммах хлоридов в 1 л воды: 1 мг-экв Cl- = 35,453 Cl-; 1 мг Cl- = 0,0282 мг-экв Cl-.
Качественное определение
47.2. Приблизительно 10 мл пробы в пробирке окисляют несколькими каплями азотной кислоты (1:4) и приливают около 0,5 мл 5%-ного раствора нитрата серебра. В зависимости от концентрации хлоридов возникают опалесценция, мутность или осадок. Последние растворяют избытком аммиака.
Аргентометрическое определение по Мору
47.3. Хлорид-ионы осаждают титрованным раствором нитрата серебра в нейтральной или слабощелочной среде  в виде малорастворимого хлорида серебра. (Произведение растворимости хлорида серебра при 25 °C составляет 1,56·10-10.) В качестве индикатора применяют раствор хромата калия, который реагирует с избыточными ионами серебра, вызывая переход лимонно-желтой окраски в оранжево-желтую. При определении можно пользоваться и потенциометром с серебряным и каломельным электродами. Титруемый раствор соединяется с электродом сравнения при помощи солевого мостика с раствором нитрата калия.
в виде малорастворимого хлорида серебра. (Произведение растворимости хлорида серебра при 25 °C составляет 1,56·10-10.) В качестве индикатора применяют раствор хромата калия, который реагирует с избыточными ионами серебра, вызывая переход лимонно-желтой окраски в оранжево-желтую. При определении можно пользоваться и потенциометром с серебряным и каломельным электродами. Титруемый раствор соединяется с электродом сравнения при помощи солевого мостика с раствором нитрата калия.
в виде малорастворимого хлорида серебра. (Произведение растворимости хлорида серебра при 25 °C составляет 1,56·10-10.) В качестве индикатора применяют раствор хромата калия, который реагирует с избыточными ионами серебра, вызывая переход лимонно-желтой окраски в оранжево-желтую. При определении можно пользоваться и потенциометром с серебряным и каломельным электродами. Титруемый раствор соединяется с электродом сравнения при помощи солевого мостика с раствором нитрата калия.Метод применяется для определения хлоридов при содержании их более 2 мг/л; без разбавления можно титровать пробы с содержанием хлоридов до 400 мг/л. Точность определения составляет +/- 1 - 3 мг/л. Для точного определения хлоридов при концентрациях менее 10 мг/л пробы необходимо упаривать. В зависимости от концентрации хлоридов в пробе используют 0,1 н.; 0,05 н. или 0,02 н. титрованные растворы нитрата серебра.
Мешающие влияния
47.4. Окраску прозрачных проб устраняют встряхиванием 100 мл пробы с примерно 0,5 г активного угля (препарат не должен содержать хлоридов, что устанавливается холостым опытом с дистиллированной водой). После обесцвечивания пробу фильтруют через плотный бумажный фильтр (синяя лента) и фильтр промывают дистиллированной водой.
При наличии мутности и окраски, мешающих определению, пробу осветляют суспензией гидроокиси алюминия или же обесцвечивают активным углем. К 100 мл пробы прибавляют 3 мл суспензии гидроокиси алюминия и смесь встряхивают до обесцвечивания жидкости. Затем пробу фильтруют через фильтр "белая лента" и осадок промывают дистиллированной водой. Хлориды определяют во всем фильтрате. (Приготовление суспензии гидроокиси алюминия описано на стр. 177).
Аргентометрическим методом определяются вместе с хлоридами бромиды, йодиды и цианиды. Цианиды разрушают перекисью водорода в щелочной среде, бромиды и йодиды в природных водах обычно отсутствуют.
Определению мешают сульфиты, сульфиды и тиосульфаты. Сульфиты устраняют прибавлением перекиси водорода к нейтральной пробе. Сульфиды и тиосульфаты разлагают перекисью водорода в щелочной среде.
Фосфаты при концентрациях выше 10 мг/л мешают точному определению точки эквивалентности.
Реактивы
47.5. Дважды дистиллированная вода.
Серная кислота, приблизительно 1 н. раствор: 28 мл концентрированной H2SO4 ч.д.а. разбавляют дистиллированной водой до 1 л.
Едкий натр, приблизительно 1 н. раствор: 40 г NaOH ч.д.а. растворяют в дважды дистиллированной воде и доводят до 1 л.
Фенолфталеин, 0,5%-ный раствор, приготовление см. на стр. 36.
Хромат калия, 5%-ный раствор: 50 г KCrO4 ч.д.а. растворяют в небольшом объеме дважды дистиллированной воды и прибавляют раствор нитрата серебра до начала образования красного осадка. После двухчасового отстаивания раствор фильтруют и доводят дважды дистиллированной водой до 1 л.
Нитрат серебра, 0,1 н. (0,05 н. и 0,02 н.): 16,9874 г (8,4937 г или 3,3975 г) AgNO3 ч.д.а., высушенного при 105 °C, растворяют в дважды дистиллированной воде и доводят до 1 л. Титр или поправку раствора определяют титрованием 5 мл основного раствора хлорида натрия соответствующей нормальности, разбавленного до 100 мл дважды дистиллированной водой. Далее титруют описанным ниже методом при помощи микробюретки.
Хлорид натрия, 0,1 н. (0,05 н. и 0,02 н.) основной раствор: 5,8443 г (2,9221 г или 1,1683 г) NaCl ч.д.а., высушенного при 105 °C, растворяют в дважды дистиллированной воде и доводят до 1 л при 20 °C.
Ход определения
47.6. Для определения берут 100 мл фильтрованной пробы или меньшее ее количество и доводят до 100 мл дважды дистиллированной водой. Кислые и щелочные пробы нейтрализуют едким натром или серной кислотой по фенолфталеину. Пробы, pH которых находится в пределах значений 7 - 10, предварительно не подготавливают. Затем к пробе прибавляют 1 мл раствора хромата калия и при постоянном перемешивании титруют раствором нитрата серебра до начала перехода лимонно-желтой окраски в оранжево-желтую.
Так же проводят холостое определение с дважды дистиллированной водой.
Расчет. Содержание хлорид-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:

ИС МЕГАНОРМ: примечание. Формула дана в соответствии с официальным текстом документа. |

где a - расход раствора нитрата серебра при титровании пробы, мл;
b - расход раствора нитрата серебра при холостом определении, мл;
N - нормальность использованного титрованного раствора;
k - поправка для приведения концентрации раствора нитрата серебра к заданной нормальности;
V - объем пробы, взятой для определения, мл;
34,45 - эквивалент хлорид-иона.
Округление результатов
Диапазон, мг/л | 1,0 - 10,0 <*> | 10 - 50 | 50 - 100 | 100 - 200 | 200 - 500 |
Округление, мг/л | 0,1 | 1 | 2 | 5 | 10 |
--------------------------------
Меркурометрическое определение
47.7. Низкая константа диссоциации хлорида ртути (II) (2,6·10-15) позволяет достаточно точно определять хлорид-ионы при титровании раствором нитрата ртути (II). Точку эквивалентности определяют дифенилкарбазоном, который вместе с избытком ионов ртути образует соединение фиолетовой окраски. Титрование производят при pH = 2,5 +/- 0,1.
Без разбавления в объеме 100 мл пробы можно определить хлориды в концентрациях до 100 мг/л. Точность определения +/- 1 мг/л. Для точного определения хлоридов при концентрациях менее 10 мг/л пробу необходимо упаривать.
Мешающие влияния
47.8. Резкость перехода окраски индикатора в значительной степени зависит от концентрации водородных ионов. В растворах с pH = 2,0 индикатор не окрашивается, при pH = 3,0 возникновение окраски запаздывает. Точное установление pH предусмотрено в ходе анализа.
Определению мешают сульфиты, хроматы и трехвалентное железо в концентрациях более 10 мг/л.
Вместе с хлоридами этим методом определяются йодиды и бромиды, присутствие которых в обычных условиях не предполагается.
Мешающее влияние сульфидов, цианидов большой мутности и окраски устраняют так же, как и при аргентометрическом определении.
Реактивы
47.9. Дважды дистиллированная вода.
Азотная кислота, примерно 0,2 н. раствор: 12,7 мл концентрированной HNO3 ч.д.а. без хлоридов разбавляют дважды дистиллированной водой до 1 л.
Едкий натр, 0,1 н. раствор: 4 г NaOH ч.д.а. растворяют в 1 л дважды дистиллированной воды.
Смешанный индикатор: 0,5 г дифенилкарбазона ч.д.а. и 0,05 г бромфенолового синего растворяют в 100 мл 95%-ного этилового спирта. Раствор сохраняют в темной бутылке.
Нитрат ртути (II), 0,05 н. раствор: 8,5 г Hg(NO3)2·1/2H2O ч.д.а. увлажняют 1 мл концентрированной азотной кислоты ч.д.а., растворяют в небольшом количестве воды и доводят до 1 л. (Для приготовления раствора можно применить также окись ртути; 5,5 г HgO ч.д.а. растворяют в небольшом избытке концентрированной азотной кислоты ч.д.а.) Поправку к титру определяют титрованием 5 мл 0,05 н. основного раствора хлорида натрия, доведенного до 100 мл дважды дистиллированной водой. Титрование проводят, как и при анализе пробы.
Хлорид натрия, 0,05 н. основной раствор (приготовление см. на стр. 234).
Ход определения
47.10. Для работы берут 100 мл профильтрованной пробы или меньшее количество и доводят до 100 мл дистиллированной водой. В колбе для титрования к пробе прибавляют 0,3 мл раствора индикатора, далее по каплям добавляют раствор 0,2 н. азотной кислоты до перехода окраски из сине-зеленой в желтую и сверх того еще 0,25 мл кислоты. Для сильнокислых проб, окрашивающихся после прибавления индикатора в желтый цвет, необходимо прибавить 0,1 н. едкий натр до появления сине-зеленой окраски. Затем пробу подкисляют азотной кислотой, как указано выше.
После этого пробу титруют раствором нитрата ртути (II) до перехода желтой окраски в фиолетовую.
Количество раствора нитрата ртути (II), необходимое для изменения окраски, определяют холостым опытом в 100 мл дважды дистиллированной воды.
Расчет. Содержание хлорид-ионов в мг/л (x) или в мг-экв/л (y) вычисляют по формулам:
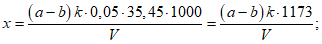

где a - расход 0,05 н. раствора нитрата ртути (II) на титрование пробы, мл;
b - расход 0,05 н. раствора нитрата ртути (II) на титрование холостой пробы, мл;
k - поправка для приведения нормальности раствора нитрата ртути (II);
V - объем пробы, взятой для определения, мл;
35,45 - эквивалент хлорид-иона;
0,05 - нормальность раствора нитрата ртути (II).
Округление результатов. Результаты округляют, как и при аргентометрическом определении.
Определение содержания малых количеств хлоридов
нефелометрическим методом
47.11. В сильно разбавленных растворах хлоридов в результате реакций между ионами хлора и серебра возникает муть, не оседающая в течение продолжительного времени.
Сравнивая мутность полученной суспензии с мутностью стандартных растворов, находят концентрации хлоридов в пробе воды. Метод применим для определения концентрации хлоридов в водных растворах, содержащих хлорид-ион в количествах не более 1,0 - 1,5 мг/л.
Аппаратура
47.12. Колориметрические пробирки из бесцветного стекла высотой 150 мм, диаметром 18 - 20 мм с притертыми пробками и плоским дном.
Микропипетка емкостью 1 мл с делениями через 0,01 мл.
Реактивы
47.13. Хлорид натрия, стандартный раствор - 0,1648 г хлорида натрия (х.ч.), предварительно перекристаллизованного и прокаленного в муфеле, растворяют в дистиллированной воде, не содержащей хлорид-иона, и доводят объем раствора до 1 л.
Полученный раствор содержит 0,1 мг/л Cl-. Раствор разбавляют в 100 раз, т.е. берут пипеткой 1,0 мл его, вливают в мерную колбу и доводят дистиллированной водой до объема 100 мл. В 1 мл полученного раствора содержится 0,001 мг Cl-.
Нитрат серебра, стандартный раствор: отвешивают 2 г нитрата серебра, помещают в мерную колбу емкостью 1 л, вливают в колбу 500 мл дистиллированной воды и 160 мл концентрированной азотной кислоты (плотность 1,4). Содержимое колбы перемешивают и охлаждают; после этого объем раствора доводится дистиллированной водой до 1 л.
Азотная кислота, плотность 1,4.
Стандартная шкала: в колориметрические пробирки с помощью бюретки вводят соответствующие количества разбавленного стандартного раствора хлорида натрия. Объем раствора в пробирках доводят дистиллированной водой до 10 мл, после чего в каждую пробирку вводят микропипеткой по 0,25 мл стандартного раствора нитрата серебра.
Растворы в пробирках шкалы соответствуют содержанию хлоридов, указанному в табл. 11.
Таблица 11
Количество стандартного раствора хлорида натрия, мл | Количество дистиллированной воды, мл | Содержание хлоридов, мг/л |
0,6 | 9,4 | 0,06 |
1 | 9 | 0,1 |
2 | 8 | 0,2 |
3 | 7 | 0,3 |
4 | 6 | 0,4 |
5 | 5 | 0,5 |
6 | 4 | 0,6 |
7 | 3 | 0,7 |
8 | 2 | 0,8 |
9 | 1 | 0,9 |
10 | 0 | 1 |
Ход определения
47.14. В колориметрическую пробирку вливают 10 мл анализируемой воды, добавляют микропипеткой 0,25 мл раствора нитрата серебра. Содержимое перемешивают встряхиванием пробирки, закрытой притертой пробкой. Дают стоять 10 мин, после чего сравнивают помутнение жидкости со шкалой стандартов, подложив под пробирки черную бумагу, и смотрят в них сверху. Метод очень чувствителен, он позволяет открыть 0,02 мг/л хлорид-иона.
Общие положения
48.1. В поверхностные воды цианиды могут попадать только при их загрязнении промышленными сточными водами.
Вследствие большой токсичности цианидов требуется очень точное их определение при самых малых концентрациях их в воде. Выполнение этой задачи затрудняется тем, что кроме простых цианидов (CN-) сточные воды содержат и комплексные цианиды, часть которых очень токсична, как, например, [Cu(CN)2]-, [Cu(CN)3]2-, [Zn(CN)4]2- и других, а часть в малых концентрациях безвредна, например [Fe(CN)6]4-.
Поскольку в загрязненной природной воде цианиды могут быть в самых малых концентрациях, предлагается один из самых чувствительных и быстрых методов для их определения - метод с применением пиридин-бензидинового реактива. Этот метод дает возможность определить 0,025 мг CN- в 1 л воды. Пробы вод, предназначенные для определения в них содержания цианидов, необходимо консервировать едким натром, доводя pH до величины не менее 11, если эти пробы не анализируют непосредственно после их отбора.
Определение цианидов с бензидином и пиридином
48.2. Определение основано на превращении цианида (а также и роданида) в бромциан добавлением бромной воды к анализируемому нейтральному или слабокислому раствору. Бромистый циан, реагируя с бензидином и пиридином, образует окрашенные соединения производного глутаконового альдегида или так называемого полиметинового красителя, имеющего в растворе интенсивно-красную окраску (в малых концентрациях окраска розовая).
Этим методом определяются как простые цианиды, так и цианиды, связанные в комплексы с медью, цинком и другими металлами, кроме гексацианоферратов (II) и (III), которые не мешают определению при концентрациях, не превышающих 10 мг/л, независимо от содержания простых цианидов и других комплексных цианидов.
Если в воде присутствуют роданиды, они определяются вместе с цианидами. В этом случае цианиды определяют по разности между общим содержанием цианидов и роданидов и содержанием роданидов после удаления синильной кислоты из подкисленного и прокипяченного раствора. При расчетах следует помнить, что 2,2 мкг роданид-иона дают такую же окраску, как 1 мкг цианид-иона.
Поскольку в природной воде мешающие определению вещества в заметных количествах отсутствуют, метод применим без предварительной перегонки пробы. В ходе определения производят добавки бромной воды и мышьяковистой кислоты, которые уничтожают малые количества окислителей и восстановителей. Фенолы не мешают определению, так как образовавшиеся в ходе реакции бромзамещенные фенолы легко разрушаются добавлением восстановителя (мышьяковистой кислоты).
Аппаратура
48.3. Фотометр, светофильтр  .
.
.Кюветы с толщиной слоя 1 см.
Градуированные плоскодонные пробирки с притертой пробкой.
Реактивы
48.4. Бромная вода, дистиллированную воду насыщают бромом.
Мышьяковистая кислота, 2%-ный раствор. Приготовляется растворением соответствующего количества As2O3 в воде при кипячении с обратным холодильником.
Пиридиновый реактив. Смешивают 60 мл чистого пиридина (температура кипения 114 °C) с 40 мл воды и 10 мл концентрированной соляной кислоты.
Бензидин солянокислый, 5%-ный раствор в разбавленной (2:98) соляной кислоте.
Соляная кислота, 0,1 н. раствор.
н-Бутиловый или н-амиловый спирт.
Арсенит натрия, 2%-ный раствор.
Цианид калия, стандартный раствор: 0,2503 г цианида калия растворяют в 100 мл прокипяченной дистиллированной воды; 1 мл полученного раствора содержит 1 мг CN-. Перед построением калибровочной кривой этот раствор разбавляют в 100 раз и получают раствор, в 1 мл которого содержится 1 мкг CN--ионов.
Точную концентрацию стандартного раствора цианида устанавливают каждый раз перед построением калибровочной кривой аргентометрическим титрованием с индикатором Файгля (п-диметиламинобензолиденроданин). Отмеривают 10 - 25 мл стандартного раствора цианида калия в колбу для титрования, доливают дистиллированной водой приблизительно до 100 мл, подщелачивают 2 мл 10%-ной едкой щелочи, вносят 1 мл 0,03%-ного раствора индикатора Файгля в ацетоне и титруют 0,05 н. или 0,1 н. раствором нитрата серебра. Концентрацию цианидов (мг/л) в стандартном растворе рассчитывают по формуле
m = (a - b)kN·0,052,
где a - расход 0,05 н. титрованного раствора AgNO3, мл;
b - расход того же реактива на холостое определение, мл;
k - поправка для приведения концентрации раствора к расчетной;
0,052 - коэффициент пересчета на CN-;
N - нормальность титрованного раствора AgNO3.
Калибровочная кривая
48.5. Для построения калибровочной кривой прибавляют все указанные реактивы к стандартным растворам цианида калия, в 2 мл которого содержится от 0,05 до 2 мкг CN-, и измеряют оптическую плотность полученных растворов. Шкала устойчива в течение 30 мин.
Ход определения
ИС МЕГАНОРМ: примечание. Сноски даны в соответствии с официальным текстом документа. |
48.6. В градуированную плоскодонную пробирку емкостью 10 мл, снабженную притертой пробкой, вносят 2 мл анализируемой нейтральной или слабокислой испытуемой воды <*>, содержащей цианиды (и роданиды) в концентрации от 0,025 до 1 мг/л. Если концентрация цианидов и роданидов превышает предельную, воду предварительно разбавляют. Прибавляют 0,2 мл бромной воды и перемешивают. Избыток брома удаляют, добавляя 0,2 мл раствора мышьяковистой кислоты <*>. Содержимое пробирки хорошо перемешивают и из другой пробирки (или маленького цилиндрика) приливают смесь 3 мл пиридинового реактива с 0,6 мл раствора солянокислого бензидина. Жидкость снова хорошо перемешивают и через 15 - 20 мин находят оптическую плотность полученного раствора.
--------------------------------
<*> Если испытуемая вода имеет щелочную реакцию, ее предварительно нейтрализуют 0,1 н. раствором кислоты. Требуемое количество кислоты определяют, титруя другую порцию воды (2 мл) с индикатором метиловым оранжевым.
<*> Вместо мышьяковистой кислоты можно применять 0,5%-ный раствор солянокислого или сернокислого гидразина. Гидразин добавляют по каплям до исчезновения окраски брома и сверх того добавляют еще одну каплю. Большого избытка следует избегать; обычно расходуется 0,25 - 0,35 мл указанного раствора.
Расчет. Содержание цианид-ионов (x) в мг/л вычисляют по формуле

где a - содержание CN-, найденное по калибровочной кривой, мг;
V - объем пробы, взятой для анализа, мл.
Определение цианидов в присутствии роданидов
48.7. Если в воде имеются цианиды и роданиды, то определяют оптическую плотность для суммы цианидов и роданидов, затем после удаления синильной кислоты кипячением из подкисленного раствора проводят реакцию уже только для одних роданидов и снова определяют оптическую плотность.
Далее вычитают из оптической плотности для суммы цианидов и роданидов оптическую плотность для роданидов, разность приходится на цианиды. По калибровочной кривой для цианидов определяют их содержание в воде. Приведенные варианты метода: для прозрачных неокрашенных и для мутных или окрашенных проб воды.
Ход определения
48.8. Вариант А (для прозрачных неокрашенных проб воды). Параллельно в две пробирки с притертыми пробками отбирают по 2 мл анализируемой воды. В одной пробе определяют сумму цианидов и роданидов точно так, как при определении цианидов, а в другой пробе определяют роданиды. Для этого раствор подкисляют 0,1 н. HCl с таким расчетом, чтобы после нейтрализации был прибавлен избыток ее в 1 - 2 капли.
Требуемое количество кислоты находят титрованием отдельной пробы раствора 0,1 н. HCl с индикатором метиловым оранжевым. Затем помещают открытую пробирку в стакан с водой, нагревают воду до кипения и оставляют пробирку в кипящей бане на 30 мин. После этого содержимое пробирки охлаждают, вливают в нее дистиллированную воду до объема 2 мл, чтобы заменить испарившуюся при нагревании воду и добавляют все реактивы, какие применяются для определения цианидов этим методом.
Вариант Б (проба воды мутная или окрашенная). Если окраска анализируемой воды или ее мутность мешают определению, то поступают следующим образом: нейтрализуют 0,1 н. раствором HCl 5 мл испытуемой воды, содержащей 0,05 - 0,3 мкг цианид-ионов, как указано выше, и прибавляют по каплям бромную воду до появления слабой желтой окраски свободного брома. Затем приливают по каплям 2%-ный раствор арсенита натрия до исчезновения окраски брома и 1 каплю избытка. Прибавляют 5 мл п-бутилового или п-амилового спирта, закрывают пробкой и взбалтывают. Затем приливают смесь растворов пиридина и бензидина, как указано выше (стр. 240), закрывают пробкой и сильно взбалтывают. Дают постоять не менее 15 мин, отделяют слой органического растворителя и измеряют его оптическую плотность по отношению к раствору, полученному в "холостом" опыте, пользуясь светофильтром  .
.
.В этом варианте чувствительность метода возрастает. Оптимальные концентрации цианид-ионов от 0,02 до 0,5 мг/л.
Расчет. Вариант А. Для расчета содержания роданид-ионов пользуются калибровочной кривой, приготовленной по стандартным растворам роданида или же по калибровочной кривой для определения цианидов; в последнем случае полученный результат умножают на коэффициент пересчета, равный 2,2. Расчетная формула приведена на стр. 240.
Вариант Б. Расчетную форму см. на стр. 240.
Общие положения
49.1. Экстрагируемыми называются вещества, извлекаемые при соблюдении известных условий из пробы воды органическим растворителем и остающиеся после его удаления. К этой группе относятся масла, жиры, мыла, смолы, воски, тяжелые углеводороды, некоторые метаболиты планктона и др. Поскольку селективные органические растворители, в которые переходили бы отдельные типы указанных веществ, не известны, определяют суммарно содержание всех этих веществ. В зависимости от происхождения пробы воды устанавливают тип присутствующих в ней экстрагируемых веществ и выбирают наиболее подходящий метод определения.
Вследствие того, что в описываемых методах предусматривают взвешивание экстрагированного вещества после отгонки растворителя и высушивания остатка при 105 °C (за исключением определения смол), в результаты анализов не входит содержание экстрагируемых веществ с более низкой температурой кипения, а также те фракции этих веществ, которые обладают летучестью при температуре сушки.
Найденное количество экстрагируемых веществ зависит также от свойств примененного растворителя, от продолжительности и способа экстракции и от ряда других условий, поэтому для получения сравнимых результатов необходимо строго соблюдать указанный порядок работы.
Минеральные масла содержатся в сточных водах почти всех промышленных предприятий. В реках они присутствуют обычно в форме эмульсий или коллоидных растворов. Сточные воды, сбрасываемые шерстопрядильными, жироперерабатывающими предприятиями, бойнями, молочными заводами, а также городские канализационные стоки содержат жиры. Под понятием "жиры" подразумеваются все извлекаемые органическими растворителями вещества, т.е. не только глицериды и жирные кислоты, но и ароматические и алифатические углеводороды.
Ниже приводятся четыре метода определения экстрагируемых веществ.
Экстрагируемые вещества в сточных и в очень загрязненных поверхностных водах, содержащих преимущественно минеральные масла, определяют весовым методом после соосаждения с гидроокисью алюминия и экстракции петролейным эфиром.
Экстрагируемые вещества, образуемые преимущественно жирами, определяют извлечением их эфиром из упаренной пробы воды. Определение всех жиров в сумме производится экстрагированием после разложения минеральной кислотой мыл и сложных эфиров. Экстрагированием пробы без предварительного разложения определяются так называемые "непосредственно экстрагируемые жиры". По разности результатов обоих определений можно ориентировочно судить о содержании мыл.
Смолы в фенольных сточных водах определяют экстракцией эфиром.
Основным условием получения правильного результата является отбор наиболее типичной пробы. При хранении пробы воды в бутыли физические свойства экстрагируемых веществ так изменяются, что пробу нельзя уже усреднить для отбора части ее для анализа. Поэтому для определения экстрагируемых веществ пробу прямо на месте следует отбирать в особую бутыль. При анализе надо стараться, чтобы во взятую для анализа пробу попали все частицы, приставшие к стенкам сосуда. Эти частицы снимают со стенок механическим путем и ополаскиванием соответствующим растворителем.
Результаты определения приводятся в миллиграммах экстрагируемых веществ на 1 л воды с указанием на примененный метод определения.
Качественное определение
49.2. Вода, содержащая масла, обыкновенно несколько опалесцирует. Вода с содержанием жиров бывает обычно беловато-мутной. Жир может выделиться на стенках бутыли. Доказательством его присутствия является осветление воды после встряхивания ее с добавкой эфира. Если в пробе присутствует мыло, то осветление произойдет только после предварительного подкисления минеральной кислотой.
Следы масел или жиров легко обнаруживаются чувствительной пробой с камфарой: кусочек камфары (или парафенилендиамина) величиной с рисовое зернышко бросают на поверхность пробы. При отсутствии в ней масла зерно быстро перемещается по поверхности жидкости. В присутствии масла оно остается на том же месте, куда было брошено.
Определение минеральных масел
49.3. Определение основано на соосаждении масел с гидроокисью алюминия. Из раствора, получаемого после выделения и растворения осадка, экстрагируют масла петролейным эфиром, а после отгонки последнего из экстракта и высушивания оставшиеся экстрагируемые вещества взвешивают.
Аппаратура
49.4. Делительные воронки емкостью 250 мл.
Установка для перегонки с электрической водяной баней.
Сушильный шкаф (105 °C).
Реактивы
49.5. Сульфат алюминия, 10%-ный раствор: 10 г Al2(SO4)3 в расчете на безводный растворяют в дистиллированной воде и доводят до 100 мл.
Карбонат натрия, 20%-ный раствор: 20 г Na2CO3 ч.д.а. растворяют в дистиллированной воде и доводят до 100 мл.
Соляная кислота, разбавленный (1:1) раствор.
Сульфат натрия безводный, свежепрокаленный, с крупностью зерен 0,2 - 0,5 мм.
Петролейный эфир, не содержащий жира, обезвоженный безводным сульфатом натрия (точка кипения ниже 65 °C).
Ход определения
49.6. Для определения берут объем пробы с таким расчетом, чтобы в нем содержалось 50 - 2000 мг экстрагируемых веществ. На каждый литр пробы приливают 3 мл раствора сульфата алюминия и хорошо перемешивают. Затем на каждый литр смеси прибавляют 1 мл раствора карбоната натрия. Снова перемешивают и осадку с выделенными маслами дают осесть на дно сосуда. Воду над осадком сифонируют, а осадок с остатком воды переносят в меньший сосуд, где опять раствору дают отстояться, после чего снова сифонируют. Затем осадок растворяют в разбавленной соляной кислоте, переливают раствор в делительную воронку и не меньше трех раз экстрагируют петролейным эфиром (на каждую экстракцию расходуют около 20 мл). Эфирные вытяжки соединяют, промывают три раза водой и оставляют стоять в течение ночи с добавкой безводного сульфата натрия. Смесь фильтруют через сухую фильтровальную бумагу, собирая фильтрат во взвешенную колбу со шлифом емкостью примерно 100 мл. Остаток промывают на фильтре безводным петролейным эфиром. Содержащую экстракт колбу со шлифом присоединяют к холодильнику и отгоняют петролейный эфир на водяной бане. Остаток в колбе сушат 1 ч при 105 °C. После охлаждения колбы ее взвешивают.
Расчет. Содержание экстрагируемых веществ (минеральных масел) в мг/л вычисляют по формуле
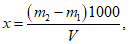
где m1 - вес пустой колбы, мг;
m2 - вес колбы с сухими экстрагируемыми веществами, мг;
V - объем пробы, взятой для определения, мл.
Округление результатов. Результаты округляют до целых миллиграммов.
Определение "общих жиров"
49.7. Определение основано на извлечении эфиром жиров из пробы воды, выпаренной с добавлением фосфорной кислоты.
После отгонки из экстракта эфира экстрагированные вещества (углеводороды и жирные кислоты) взвешивают.
Аппаратура
49.8. Сушильный шкаф (105 °C).
Аппарат Сокслета для экстрагирования.
Экстракционные патроны.
Электрическая водяная баня.
Реактивы
49.9. Ортофосфорная кислота, разбавленный (1:6) раствор.
Речной песок.
Этиловый эфир, обезвоженный добавкой безводного сульфата натрия.
Ход определения
49.10. В зависимости от предполагаемого содержания жиров отмеривают 500 мл или больший объем пробы, который выпаривают на водяной бане до меньшего объема. Затем вносят 20 - 30 г прокаленного речного песка и такое количество разбавленной фосфорной кислоты, при котором реакция смеси становится слабокислой. Смесь сушат на водяной бане, после чего еще около 1 ч в сушильном шкафу при температуре 105 °C. Высушенную смесь количественно переносят в экстракционный патрон. Последние ее остатки стирают куском фильтровальной бумаги, который помещают в тот же патрон с пробой. После этого патрон со смесью закрывают ватной пробкой, не содержащей жиров, и вставляют в цилиндрическую часть аппарата Сокслета. Во взвешенную колбу со шлифом отмеривают нужное в соответствии с размерами прибора количество безводного эфира, присоединяют колбу к прибору и ведут экстрагирование на электрической водяной бане в течение 3 - 4 ч. Затем колбу с экстрактом соединяют с холодильником, эфир отгоняют. Остаток в колбе сушат 1 ч при 105 °C и вновь взвешивают.
Расчет. Содержание экстрагируемых веществ (x) "общие жиры" в мг/л вычисляют по формуле

где m1 - вес пустой сухой колбы, мг;
m2 - вес той же колбы с высушенными экстрагированными веществами, мг;
V - объем пробы воды, взятый для определения, мл.
Округление результатов. Результаты округляются до целых миллиграммов.
Определение "непосредственно экстрагируемых жиров"
49.11. Этим методом определяют экстрагируемые вещества без омыленной части жирных кислот. Порядок определения такой же, как при определении "общих жиров", с той только разницей, что пробу не подкисляют фосфорной кислотой.
Определение смол
49.12. Смолы выделяют подкислением пробы до изоэлектрической точки (pH от 3 до 4). Выделенные смолы экстрагируют эфиром. Результат двух параллельных определений одной и той же пробы не должен отличаться от средней арифметической величины более чем на +/- 5%.
Аппаратура
49.13. Делительная воронка емкостью 250 или 500 мл с короткой трубкой.
Сушильный шкаф (42 °C).
Реактивы
49.14. Этиловый спирт ч.д.а.
Серная кислота, разбавленный (1:4) раствор.
Сульфат натрия безводный, свежепрокаленный, с крупностью зерен 0,2 - 0,5 мм.
Ход определения
49.15. В делительную воронку емкостью 250 или 500 мл отмеривают 50 - 100 мл пробы. На дно воронки помещают хорошо уплотненный слой ваты толщиной около 0,5 мм. Шлифы крана и пробки смачивают дистиллированной водой. Пробу подкисляют, прибавляя 10 мл разбавленной серной кислоты, и после взбалтывания оставляют в покое до следующего дня для выпадения смолы. (Для получения ориентировочных результатов отстаивание можно заменить аэрированием в течение 15 мин).
После выделения смолы воду из делительной воронки выпускают по каплям через слой ваты и отбрасывают. Выделенные смолы остаются на стенках воронки и на вате. Остатки воды, задержавшиеся на вате или в трубке воронки, удаляют, стряхивая их резким движением (по направлению к земле) при открытом кране или же осторожно выдувая из делительной воронки. Трубку воронки осушают ватой.
Делительную воронку помещают над обыкновенной воронкой с короткой трубкой для фильтрования, в которую вводят слой ваты. На этот слой для удаления остатка влаги насыпают примерно 2 г безводного свежепрокаленного сульфата натрия. Под воронку подставляют взвешенный стакан емкостью около 100 мл. Смолу в делительной воронке растворяют добавлением небольших порций эфира (5 - 10 порций, в зависимости от количества смолы) до тех пор, пока вытекающий эфир не будет бесцветным. После этого трубку делительной воронки, воронку со слоем ваты для фильтрования и трубку ополаскивают эфиром, стекающим в подставленный стакан. Из экстракта в стакане отгоняют эфир выпариванием на водяной бане при 40 - 45 °C или же дают ему свободно испариться в теплом месте. Остаток сушат 2 ч при 42 °C и после охлаждения взвешивают.
Расчет. Содержание экстрагируемых веществ смолы (x) в мг/л вычисляют по формуле
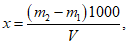
где m1 - вес пустого стакана, мг;
m2 - вес стакана с высушенными смолами, мг;
V - объем пробы, взятой для определения, мл.
Округление результатов. Результаты округляют до целых миллиграммов.
ДЛЯ ОСВЕТЛЕНИЯ И ОБЕСЦВЕЧИВАНИЯ ВОДЫ
50.1. Целью описанных в настоящем разделе технологических исследований является определение доз коагулянта, которые необходимы для удаления из исследуемой воды взвешенных и коллоидных загрязнений, вызывающих ее мутность и цветность.
Определение доз коагулянта при осветлении и обесцвечивании
воды в отстойниках и осветлителях со взвешенным осадком,
с последующим фильтрованием воды
Общие положения
50.2. Необходимые дозы коагулянта определяют:
1) для мутных вод - с предварительным подщелачиванием воды (известью) и без него;
2) для цветных вод - с предварительным хлорированием воды и без него;
3) для мутно-цветных вод - как с предварительным хлорированием и подщелачиванием, так и без них.
Требуемые количества реагентов выражают величинами наименьших доз безводного сульфата алюминия, хлора и извести, необходимых для снижения мутности воды до 10 мг/л, после отстаивания в течение 1 ч и цветности воды до 20° хромово-кобальтовой шкалы после отстаивания в течение 1 ч и последующего фильтрования воды через бумажный фильтр.
Определение ведут немедленно и во всяком случае не позднее 4 ч после отбора пробы исходной воды при температуре воды, соответствующей производственным условиям. Перед определением пробу воды тщательно перемешивают.
Ниже приведены три варианта определения: вариант А - исследуемую воду предварительно не подщелачивают и не хлорируют; вариант Б - воду предварительно подщелачивают; вариант В - воду предварительно хлорируют.
Реактивы
50.3. Сульфат алюминия (х.ч. или ч.д.а.), 1%-ный раствор: навеску в 19,5 г Al2(SO4)3·18H2O, соответствующую 10 г безводного сульфата алюминия Al2(SO4)3, помещают в мерную колбу емкостью 100 мл, растворяют при нагревании в 300 - 500 мл дистиллированной воды, охлаждают и доводят до метки дистиллированной водой.
Гидроокись кальция, 0,1%-ный раствор: 1 г предварительно прокаленной при 900 °C в течение 5 ч окиси кальция растирают в ступке и смывают кипящей дистиллированной водой в мерную колбу емкостью 1000 мл. По охлаждении (в закрытой колбе) доводят до метки дистиллированной водой и хранят, закрыв пробкой с хлоркальциевой трубкой, наполненной натронной известью. Периодически проверяют концентрацию CaO в растворе.
Хлорная вода, содержащая 0,1% свободного хлора. Хлорную воду приготовляют растворением хлорной извести в дистиллированной воде. Содержание свободного хлора определяют йодометрически или ортотолидиновым методом (см. стр. 226).
Определение необходимой дозы коагулянта
50.4. Вариант А (определение без подщелачивания и хлорирования воды).
В 6 - 10 мерных цилиндров емкостью 0,5 или 1 л наливают исследуемую воду до метки, затем в цилиндры добавляют различное количество миллилитров раствора сульфата алюминия из расчета получения различных его доз в диапазоне, охватывающем предполагаемую оптимальную дозу.
Содержимое во всех цилиндрах смешивают быстрым вращением палочки в течение 15 - 20 сек, затем в течение 3 - 5 мин пробу медленно перемешивают той же палочкой (30 - 40 об/мин) и оставляют для отстаивания, наблюдая за процессами образования хлопьев и их осаждения. Через 30 мин из каждого цилиндра пипеткой или сифоном отбирают по 200 мл воды из верхнего слоя, не взмучивая осадка, и определяют содержание в этой воде взвешенных веществ (см. стр. 25).
Затем в этой же пробе определяют цветность (см. стр. 28). По полученным данным определяют наименьшую (оптимальную) дозу безводного сульфата алюминия, снизившую мутность и цветность исследуемой воды до требуемых пределов. Затем рассчитывают дозу глинозема сернокислого (коагулянта) в мг/л по следующей формуле:

где a - оптимальная доза коагулянта по Al2(SO4)3, мг/л;
b - содержание Al2O3 в глиноземе сернокислом, %.
50.5. Вариант Б (определение с предварительным подщелачиванием исследуемой воды).
В случае неудовлетворительного хлопьеобразования при введении в воду сульфата алюминия или когда при близком к оптимальному количеству сульфата алюминия щелочность воды в пробе снижается до величины менее 0,3 - 0,5 мг-экв/л, требуется подщелачивать воду.
Дозу извести определяют проводя пробное коагулирование сульфатом алюминия, но на воде, предварительно обработанной известью. Определение проводят, как описано в варианте А. Выполняют несколько серий опытов с различными дозами извести в диапазоне, охватывающем предполагаемую оптимальную ее дозу. Оптимальной называют наименьшую дозу извести, обеспечивающую высокий эффект осветления и обесцвечивания воды, обработанной известью и сульфатом алюминия.
По полученным данным определяют требуемое количество гидроокиси кальция. Необходимую дозу технической извести в мг/л рассчитывают по следующей формуле:

где a - доза гидроокиси кальция, выраженная в CaO, мг/л;
b - содержание CaO в технической извести, %.
50.6. Вариант В (определение с предварительным хлорированием исследуемой воды). Определение проводят так же, как и при определении дозы коагулянта с подщелачиванием, но вместо раствора гидроокиси кальция в каждый цилиндр добавляют в первой серии опытов 1 мл хлорной воды, во второй серии - 2 мл, в третьей серии - 3 мл и т.д. Ход определения описан в варианте А. По полученным данным определяют возможность снижения дозы коагулянта за счет предварительного хлорирования и требуемые дозы хлора и коагулянта.
Определение дозы коагулянта при контактном осветлении воды
50.7. В 10 колб или мерных цилиндров наливают по 0,5 или 1 л исследуемой воды; затем в приготовленные пробы вводят раствор коагулянта в количествах, соответствующих предполагаемому диапазону требуемых доз.
Пробы воды смешивают десятикратным опрокидыванием колб или цилиндров и сразу фильтруют через промытые бумажные фильтры типа "белая лента". В фильтрате определяют взвешенные вещества и цветность (см. стр. 25 и 28).
Результаты выражают в виде графиков зависимости мутности и цветности фильтрата от дозы коагулянта. Характерный вид графиков показан на рис. 10. По полученным данным определяют пороговую дозу коагулянта (начало участка кривой, на котором качество фильтрата практически не зависит от дозы коагулянта).

и цветности воды от дозы коагулянта
Требуемую для контактного осветления дозу коагулянта определяют умножением полученного значения дозы на коэффициент запаса, равный 1,1 - 1,15.
Примечание. В процессе эксплуатации сооружений определенные по методикам данного раздела дозы реагентов уточняют, исходя из условия получения воды требующегося качества.
51.1. Для улучшения процесса осаждения взвешенных веществ при коагулировании в воду помимо коагулянтов можно добавлять флокулянты - полиакриламид (ПАА) или активированную кремниевую кислоту (АК). Наименьшая необходимая доза флокулянта должна снижать содержание взвеси в воде, предварительно обработанной коагулянтами, до 10 мг/л (после 30 мин отстаивания).
Дозу флокулянтов определяют немедленно после отбора пробы воды, и во всяком случае не позднее 4 ч после отбора при температуре воды, соответствующей производственным условиям.
Приборы
51.2. Мешалка для приготовления раствора ПАА.
Термостат для созревания активированной кремниевой кислоты при температуре, соответствующей производственным условиям.
Реактивы
51.3. Сульфат алюминия, 1%-ный раствор (приготовление см. стр. 247).
Полиакриламид, 0,1%-ный раствор: 0,2 г ПАА (из расчета на безводный продукт) помещают в лопастную мешалку (типа используемой для размельчения тканей, 1000 об/мин), заливают 200 мл дистиллированной воды и перемешивают 20 мин до получения однородного раствора.
Активированная кремниевая кислота, 0,5%-ный раствор по SiO2 готовят следующим образом: готовят рабочие растворы жидкого стекла с концентрацией 1,7% по SiO2 и сульфата алюминия с концентрацией 2,5% по Al2(SO4)3.
Исходными продуктами для приготовления рабочих растворов служат стекло натриевое жидкое, имеющее силикатный модуль, равный 3, и глинозем сернокислый очищенный.
Силикатный модуль (M) жидкого стекла - отношение числа грамм-молекул SiO2 к числу грамм-молекул Na2O - вычисляют по формуле:

где A - содержание SiO2, %;
B - содержание Na2O, %;
1,032 - отношение молекулярного веса Na2O к молекулярному весу SiO2.
Содержание SiO2 и Na2O в исходном жидком стекле определяют по данным завода-изготовителя.
К 75 мл рабочего раствора жидкого стекла 1,7%-ного по SiO2 при интенсивном перемешивании (1000 об/мин) добавляют постепенно в течение 1 - 2 мин 25 мл рабочего раствора сульфата алюминия. Перемешивание продолжают после добавления всего количества сульфата алюминия еще 2 мин, затем полученный золь ставят для созревания на 1 ч в термостат, в котором поддерживают температуру, соответствующую производственным условиям.
По истечении 1 ч к золю добавляют 155 мл воды и тщательно перемешивают. Полученный 0,5%-ный по SiO2 раствор АК используют при проведении пробного коагулирования.
Ход определения
51.4. В 10 мерных цилиндров емкостью 0,5 или 1 л наливают исследуемую воду до метки и вводят коагулянт в количестве, соответствующем оптимальной дозе (см. стр. 248).
Содержимое всех цилиндров смешивают, как указано на стр. 248. Через 3 мин вводят во вторую и последующие пробы раствор ПАА или АК в количествах, обеспечивающих получение различных концентраций (доз) в интервале значений, близких к предполагаемой оптимальной дозе флокулянта.
Пробы с флокулянтом смешивают, затем перемешивают стеклянной палочкой (30 - 40 об/мин) в течение 3 - 5 мин и оставляют в покое, наблюдая за образованием хлопьев и осаждением взвеси. Через 30 мин из каждого цилиндра пипеткой или сифоном из верхнего слоя воды отбирают пробы по 100 - 200 мл (не взмучивая осадка).
В пробах определяют содержание взвешенных веществ (см. стр. 25) и по полученным данным устанавливают наименьшую дозу флокулянта (мг/л), достаточную для снижения содержания взвешенных веществ в воде до 10 мг/л.
Затем проводят аналогичные опыты с меньшими дозами коагулянта.
По полученным данным определяют возможность снижения дозы коагулянта за счет использования флокулянтов и устанавливают требуемые дозы коагулянта и флокулянта, исходя из условия получения требуемого эффекта очистки воды.
Примечание. В процессе эксплуатации сооружений полученную по указанной методике дозу флокулянта уточняют по данным работы фильтров. Оптимальной является доза, при которой время защитного действия равно или несколько больше времени достижения предельной потери напора в фильтрующей загрузке (см. стр. 256).
Для улучшения очистки воды на контактных осветлителях флокулянты вводят непосредственно перед этими сооружениями (но не менее чем через 2 - 3 мин после введения коагулянта). В этом случае в качестве оптимальной принимают дозу флокулянта, при которой время защитного действия загрузки равно или несколько больше времени достижения предельной потери напора в ней. Аналогично выбирают дозу флокулянтов при их непосредственной подаче перед фильтрами в двухступенчатой схеме очистки воды.
Примечание. Применение флокулянтов эффективно только в тех случаях, когда время защитного действия загрузки фильтров (или контактных осветлителей) меньше времени достижения в них предельной потери напора.
52.1. Осаждаемость взвесей <*> определяют для расчета скорости осаждения естественной или скоагулированной взвеси. Эти данные необходимы для расчета отстойников вновь проектируемых водопроводных станций, а также для оценки работы эксплуатируемых отстойников.
--------------------------------
<*> Под термином "взвесь" понимают малоустойчивую дисперсную систему, твердые частицы которой сравнительно быстро выпадают в осадок.
Осаждаемость взвесей, частицы которых не изменяют своего размера в процессе осаждения (взвесь I), можно определять с помощью лабораторных стеклянных цилиндров (вариант А) или торзионных весов (вариант Б).
Осаждаемость естественных и скоагулированных взвесей, частицы которых способны укрупняться в процессе осаждения (взвесь II), определяют с помощью высоких цилиндров  с конусным дном и конусным верхом.
с конусным дном и конусным верхом.
с конусным дном и конусным верхом.Определение осаждаемости взвесей типа I
52.2. Вариант А (определение осаждаемости в лабораторных цилиндрах). Несколько пронумерованных цилиндров 2 (рис. 11) (желательно не менее 5) емкостью 0,5 - 1 л заполняют исследуемой водой до метки и устанавливают в помещении с постоянной температурой, близкой к температуре исследуемой воды, или в проточную водяную ванну 1, в которой температура воды имеет одинаковую температуру с исследуемой водой. Пробы оставляют спокойно стоять. Время отстаивания принимают для всех цилиндров разным, возрастающим от первого цилиндра к последнему.
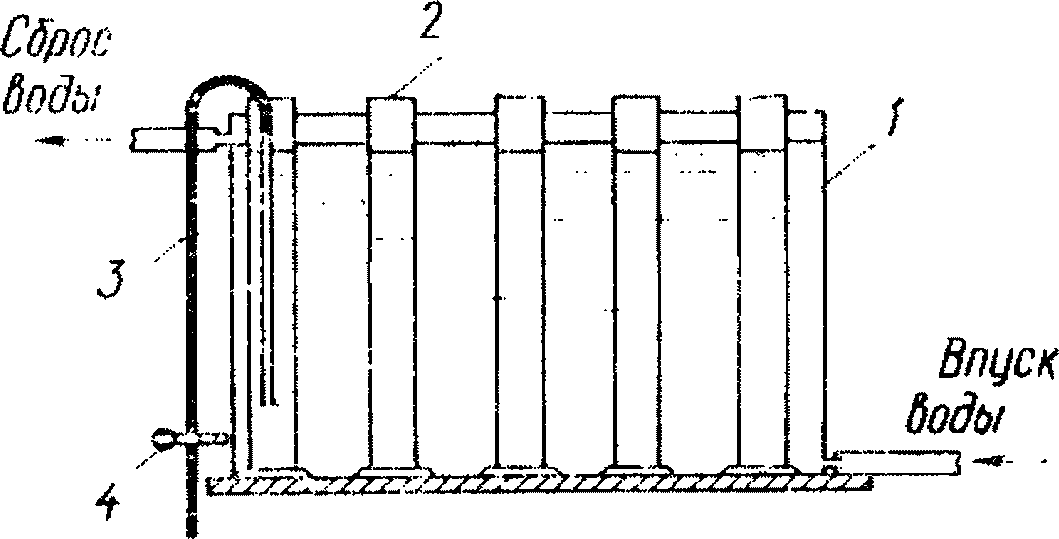
1 - ванна из органического стекла; 2 - цилиндры; 3 - сифон;
4 - зажим
Количество выпавшей в осадок взвеси в последнем цилиндре должно составлять 80 - 95% начального содержания. Промежутки времени между отбором проб при исследовании грубодисперсных суспензий принимают меньшими, при исследовании тонкодисперсных суспензий - большими.
По истечении заданного для каждого цилиндра времени отстаивания верхний слой воды на некоторую глубину (h мм) осторожно сифоном 3 сливают и определяют в этих пробах содержание взвешенных веществ (Pn).
Отношение веса выпавшего осадка к количеству взвешенных веществ в исходной воде, выраженное в процентах, характеризует осаждаемость взвеси различной крупности (An), выпадающей в осадок с высоты h за определенное время (эффект осветления воды).
Осаждаемость взвеси (в %) рассчитывают по формуле

где W - объем исследуемой воды в цилиндре в верхнем слое высотой h мм, л;
C0 - концентрация взвешенных веществ в исходной исследуемой воде, мг/л;
Pn - количество взвешенных веществ, оставшихся в слое воды высотой h после выпадения в осадок взвеси за время t сек, мг.
Результаты исследования представляют в виде графика (рис. 12). По оси абсцисс откладывают гидравлическую крупность взвеси U0 (мм/сек), определяемую делением высоты осаждения h (мм) на продолжительность отстаивания пробы t (сек), а по оси ординат - соответствующее количество выпавшей в осадок взвеси, выраженное в процентах от исходной концентрации (An).

веществ от гидравлической крупности взвеси
52.3. Вариант Б (определение осаждаемости взвеси на торзионных весах). Определение производят непосредственным взвешиванием осадка, который образуется при выпадении из воды взвешенных веществ.
Для выполнения анализа используют торзионные весы, стеклянный мерный цилиндр емкостью 0,5 л, секундомер и ручную мешалку для перемешивания исследуемой воды в цилиндре перед началом опыта (рис. 13).

на торзионных весах
1 - опора; 2 - тренога; 3 - опорные винты; 4 - шкала;
5 - указатель веса; 6 - рычаг натяжения; 7 - указатель
равновесия; 8 - тарировочная головка; 9 - коромысло;
10 - крюк; 11 - футляр; 12 - закрепительный рычаг;
13 - уровень; 14 - цилиндр; 15 - чашка; 16 - мешалка
Для взвешивания весы устанавливают по уровню 13 посредством опорных винтов 3. Затем, освободив коромысло 9 передвижением закрепительного рычага 12, устанавливают с помощью рычага натяжения 6 указатель веса 5 на нуль. После этого вращением тарировочной головки 8 совмещают указатель равновесия 7 с чертой равновесия. Точность весов периодически контролируют с помощью аналитических разновесов.
Определения производят в помещении с постоянной температурой, близкой к температуре исследуемой воды; возможно также термостатирование цилиндра путем установки его в ванне с проточной водой.
Перед анализом определяют вес чашки в дистиллированной воде при температуре, соответствующей температуре исследуемой воды (градуировку весов производят при той же температуре). Для этого под коромыслом весов устанавливают цилиндр 14 с налитой до метки дистиллированной водой. Затем осторожно, чтобы не захватить пузырьков воздуха, чашку 15 опускают в цилиндр и подвешивают к коромыслу весов. Замеряют вес чашки в воде путем перемещения указателя веса до тех пор, пока указатель равновесия не совместится с чертой равновесия, нанесенной на шкале.
После этого в цилиндр наливают исследуемую воду со взвесью. Снова опускают чашку в воду так, чтобы не захватить пузырьков воздуха (опускать боком). Опустив чашку у стенки цилиндра на глубину 1 - 2 см от уровня жидкости, держат ее с помощью пинцета в таком положении, а другой рукой с помощью мешалки 16 тщательно перемешивают содержимое цилиндра.
После окончания перемешивания мешалку вынимают, а чашку опускают на расчетную глубину h от уровня воды в цилиндре. В этот момент включают секундомер.
Расчетная глубина погружения чашки не является постоянной величиной, ее назначают в зависимости от концентрации взвешенных веществ в исследуемой воде с учетом того, чтобы объем осадка, образующийся в результате выпадения взвеси из столба жидкости над чашкой, не превышал емкость самой чашки. Поэтому для исследуемой воды большой мутности погружение чашки должно быть небольшим; для воды с невысокой концентрацией взвешенных веществ глубина погружения чашки должна быть максимально возможной.
Для построения кривой осаждения взвеси через определенные интервалы времени отсчитывают вес осадка, осевшего на чашке. Вес осадка равен разности отсчетов по шкале, полученных при взвешивании чашки с осадком и пустой чашки. Интервалы времени между замерами выбирают на основании визуального наблюдения за процессом осаждения взвеси. Последний замер веса осадка производят после того, как практически вся взвесь в слое воды над чашкой перейдет в осадок. Вес этого осадка принимают за 100%. Затем определяют веса осадков для всех ранее произведенных отсчетов и выражают их в процентах от исходной концентрации. На графике (рис. 12), по оси абсцисс которого отложена гидравлическая крупность взвеси, а по оси ординат - количество выпавшего осадка в процентах, наносят экспериментальные точки и по ним строят кривую осаждения взвеси.
Определение осаждаемости взвесей типа II
52.4. Определение производят в высоком цилиндре, в котором высота столба исследуемой жидкости может быть задана равной или соизмеримой с глубиной осадочной части производственных отстойников (рис. 14).

типа II
1 - цилиндр; 2 - кожух; 3 - бак; 4 - мешалка; 5 - патрубок
цилиндра; 6 - патрубок бака; 7 - пробка; 8 - верхняя метка;
9 - зажим; 10 - нижняя метка
Определение производят в помещении с постоянной температурой, близкой к температуре исследуемой воды, или при протоке через кожух цилиндра воды, имеющей одинаковую температуру с исследуемой водой.
Осаждаемость естественных взвесей определяют следующим образом: цилиндр 1 из органического стекла (диаметром 50 - 60 мм), имеющий с обоих концов конусы, устанавливают в вертикальном положении.
Боковой патрубок 5 в нижней части цилиндра соединяют резиновой муфтой с таким же патрубком 6 бака 3, заполняемого исследуемой водой.
Для предотвращения выпадения взвеси в осадок до начала опыта воду в баке 3 непрерывно перемешивают мешалкой 4.
Для перепуска воды из бака 3 в цилиндр 1 вынимают из соединительного патрубка резиновую пробку 7 и одновременно создают вакуум в цилиндре с помощью вакуум-насоса, подсоединенного к верхней части цилиндра. Заполнение производят до метки 8. После заполнения цилиндра 1 соединительный патрубок закрывают пробкой 7 и сразу включают секундомер. Затем отсоединяют цилиндр 1 от вакуум-насоса.
По истечении 15 - 20 мин через зажим 9 отбирают пробу воды объемом 0,1 л и потом производят отбор таких же проб каждые 20 - 30 мин. Последняя проба должна быть отобрана через 5 - 6 ч после начала опыта.
Количество проб и интервалы времени между их отборами должны быть уточнены в зависимости от скорости осаждения взвеси; для быстро осаждающейся взвеси интервалы нужно уменьшать, для медленно осаждающейся - увеличивать.
В каждой отобранной пробе определяют содержание взвеси и подсчитывают гидравлическую крупность выпавшей взвеси, а также эффект осветления (осаждаемость взвеси) для каждой продолжительности осаждения взвеси t1, t2, ..., tn по формулам:


где Un - гидравлическая крупность взвеси (мм/сек), выпадающей за время tn;
hn - высота столба воды в цилиндре в мм перед отбором n-й пробы (hn отсчитывают от нижней метки 10, отмечающей объем 0,1 л в нижней части цилиндра);
tn - время в сек, отсчитываемое от начала опыта до момента отбора n-й пробы;
An - эффект осветления воды к моменту отбора n-й пробы в % (осаждаемость взвеси);
W - объем воды в цилиндре 1 в начале опыта, л;
C0 - содержание взвеси в исходной воде, мг/л;
G1, G2, ..., Gn - вес осадка в мг в 1-й, 2-й и т.д. пробах, отобранных соответственно через промежутки времени t1, t2, ..., tn от начала опыта;
n - порядковый номер отобранной пробы.
На основе полученных данных строят график зависимости осаждаемости взвеси от гидравлической крупности выпавшей взвеси (см. рис. 12).
Осаждаемость взвесей типа II, образующихся при обработке воды реагентами, определяют таким же образом, как и осаждаемость естественных взвесей, способных к укрупнению в процессе осаждения. Перед заполнением цилиндра исследуемую воду соответствующим образом обрабатывают: вводят реагенты и перемешивают с оптимальной скоростью до образования достаточно крупных хлопьев. Затем поступают так же, как при исследовании естественной взвеси типа II.
Скорость подъема уровня жидкости в цилиндре в процессе его заполнения не должна превышать величины, при которой хлопья скоагулированной взвеси начинают разрушаться. Целесообразную продолжительность отстаивания и интервалы между отборами проб уточняют экспериментально.
И РАСЧЕТ ФИЛЬТРУЮЩИХ ЗАГРУЗОК
53.1. Определение фильтровальных характеристик воды проводят в целях получения данных, которые могут быть использованы для расчета как вновь проектируемых фильтровальных сооружений с зернистой загрузкой, так и при оценке работы эксплуатируемых фильтров для оптимизации режима их работы. Определение фильтровальных характеристик воды основано на теории технологического моделирования процесса фильтрования <*>.
--------------------------------
Экспериментальная установка
53.2. Для определения фильтровальных технологических характеристик воды необходимо иметь фильтровальную колонку  и высотой 2,5 - 3 м (рис. 15). По высоте колонки 4 устанавливают на расстоянии 20 см друг от друга пробоотборники. Пробоотборники 1 делают из тонкой медной (или латунной) трубки внутренним диаметром 2 - 3 мм. Трубка имеет штуцер, который ввинчивается в стенку фильтровальной колонки. Часть трубки, располагаемая в колонке, должна иметь длину не менее 100 мм. На этой части трубки равномерно по ее длине просверливают 10 - 12 отверстий диаметром 1 мм, после чего на нее навивают с прозорами 0,1 - 0,2 мм медную (или латунную) проволоку диаметром 1 - 1,5 мм.
и высотой 2,5 - 3 м (рис. 15). По высоте колонки 4 устанавливают на расстоянии 20 см друг от друга пробоотборники. Пробоотборники 1 делают из тонкой медной (или латунной) трубки внутренним диаметром 2 - 3 мм. Трубка имеет штуцер, который ввинчивается в стенку фильтровальной колонки. Часть трубки, располагаемая в колонке, должна иметь длину не менее 100 мм. На этой части трубки равномерно по ее длине просверливают 10 - 12 отверстий диаметром 1 мм, после чего на нее навивают с прозорами 0,1 - 0,2 мм медную (или латунную) проволоку диаметром 1 - 1,5 мм.
и высотой 2,5 - 3 м (рис. 15). По высоте колонки 4 устанавливают на расстоянии 20 см друг от друга пробоотборники. Пробоотборники 1 делают из тонкой медной (или латунной) трубки внутренним диаметром 2 - 3 мм. Трубка имеет штуцер, который ввинчивается в стенку фильтровальной колонки. Часть трубки, располагаемая в колонке, должна иметь длину не менее 100 мм. На этой части трубки равномерно по ее длине просверливают 10 - 12 отверстий диаметром 1 мм, после чего на нее навивают с прозорами 0,1 - 0,2 мм медную (или латунную) проволоку диаметром 1 - 1,5 мм.
фильтровальных характеристик воды
1 - пробоотборники; 2 - стабилизующий бачок; 3 - бачок
для исходной воды; 4 - фильтровальная колонка;
5 - пьезометры
Пробоотборник, расположенный до входа воды в загрузку, служит для контроля концентрации взвеси в исходной воде. Пробоотборник, расположенный за загрузкой, служит для контроля фильтрата. Остальные пробоотборники предназначаются для определения изменения концентрации взвеси в фильтрационном потоке (в зернистой загрузке).
Во время опыта используют не все пробоотборники, но не менее чем 6 - 7. В ходе опытов должно быть обеспечено непрерывное истечение воды из пробоотборников. Суммарный расход воды из пробоотборников не должен превышать 5% общего расхода воды, проходящей через колонку.
Колонка оснащена двумя пьезометрами 5 для определения общей потери напора по высоте загрузки.
Колонку загружают возможно более однородным зернистым материалом (песок, антрацит, керамзит и т.д.). Средний диаметр зерен загрузки должен быть в пределах от 0,7 до 1,1 мм. Отклонение в размерах наиболее крупных и наиболее мелких зерен от среднего диаметра зерен допускается в пределах 10%. Толщина слоя песка должна быть 1,5 - 2 м.
Необходимое количество загрузки (кг) рассчитывают по формуле:

где Q - вес сухого отмытого и отсортированного материала загрузки, кг;
m - пористость загрузки в реальном фильтре (для песка - 0,41 - 0,42);
W - требуемый объем загрузки, дм3.
После заполнения песок в колонке уплотняют постукиванием по стенке колонки до метки, соответствующей заданному объему загрузки (W).
Установка должна работать с постоянной скоростью фильтрования в пределах 5 - 10 м/ч.
При определении параметров работы фильтров на колонку подают воду из трубопровода, питающего производственные фильтры, или же подают воду после лабораторной установки, воспроизводящей работу отстойников или осветлителей со взвешенным осадком. Если определяют параметры фильтрования для процессов контактного осветления, то на колонку подают воду из водоисточника после добавления к ней реагентов в оптимальных количествах.
Проведение опыта
53.3. В ходе опыта в журнале наблюдений записывают результаты определений концентраций взвеси во всех пробах и потерю напора в загрузке. Концентрацию взвеси в воде при проведении технологического моделирования определяют с помощью фотоэлектроколориметра.
Частоту контрольных определений устанавливают в зависимости от хода опыта. Важно по возможности точнее определить начало ухудшения качества воды, отбираемой с различной высоты, и проследить дальнейшее изменение концентрации взвеси в пробах. При малой загрязненности фильтруемой воды и скорости фильтрования 5 - 6 м/ч достаточно отбирать пробы через каждый час. При большой загрязненности воды или более высокой скорости фильтрования может потребоваться отбор проб через каждые 30 и даже 15 мин.
В журнал записывают также характеристику качества воды источника водоснабжения и условия проведения эксперимента: крупность зерен и толщину слоя загрузки, скорость фильтрования, температуру воды.
Обработка результатов опыта и определение
параметров фильтрования
53.4. Предварительная обработка опытных данных заключается в построении "выходных" кривых для каждой точки отбора проб и кривой прироста потери напора. При правильно выбранных условиях опыта эти кривые имеют вид, показанный на рис. 16 и 17. На графике (рис. 16) проводят горизонтальную прямую с ординатой, соответствующей заданному значению величины C/C0, т.е. отношение допускаемой концентрации взвеси в фильтрате - C к исходной концентрации ее в воде, поступающей на фильтры, - C0.
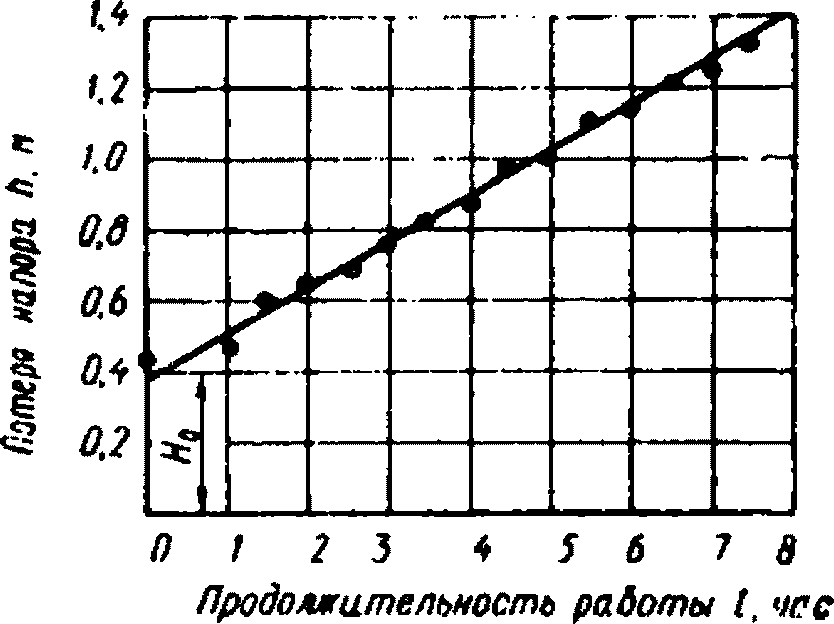
По точкам пересечения этой прямой с выходными кривыми (x) определяют продолжительность защитного действия каждого слоя загрузки tз. Эти значения откладывают на графике (рис. 18), где по оси ординат указаны толщины слоев загрузки, а по оси абсцисс - продолжительность защитного действия. По опытным точкам проводят усредненную прямую. Полученная прямая позволяет определить параметры процесса фильтрования b и a/b. Параметр b, характеризующий интенсивность прилипания взвеси к зернам загрузки и имеющий размерность м-1, определяют по формуле

x0 - отрезок, отсекаемый от оси ординат на рис. 18.

Таблица 12
при расчете фильтров
C0 | 0,03 | 0,05 | 0,10 | 0,15 | 0,20 | 0,30 | 0,40 | 0,50 |
X0 | 4,2 | 3,7 | 3,0 | 2,2 | 1,8 | 1,2 | 0,92 | 0,70 |
K | 1,86 | 1,69 | 1,51 | 1,45 | 1,36 | 1,23 | 1,10 | 1,0 |
Таблица 13
при расчете контактных осветлителей
c/c0 | 0,02 | 0,05 | 0,10 | 0,15 | 0,20 | 0,30 | 0,40 | 0,50 |
X0 | 8,2 | 6,8 | 5,6 | 4,9 | 4,3 | 2,8 | 1,90 | 0,7 |
K | 1,40 | 1,29 | 1,24 | 1,19 | 1,15 | 1,11 | 1,04 | 1,0 |
Параметр a/b имеет размерность скорости (м/ч) и характеризует скорость проникновения хлопьев (осадка) в глубь загрузки. Его значение определяют по формуле

где n - тангенс угла  наклона прямой (рис. 18);
наклона прямой (рис. 18);
Использование фильтровальных характеристик
для расчета фильтров и контактных осветлителей
53.5. Полученные значения параметров фильтрования b и a/b, а также значение среднего часового прироста потери напора h/t, определяемого с помощью пьезометров, позволяют провести расчет оптимального режима работы фильтров и контактных осветлителей. Расчет проводят по следующим формулам:



Из последней формулы вычисляют значение функции предельной насыщенности осадком порового пространства загрузки - F(A), по которому из табл. 14 определяют необходимый для дальнейших расчетов параметр A, характеризующий предельную насыщенность осадком порового пространства фильтрующей загрузки.
Таблица 14
A | 0 | 0,1 | 0,2 | 0,3 | 0,4 | 0,45 | 0,50 | 0,55 | 0,60 | 0,70 |
F(A) | 0 | 0,35 | 0,85 | 1,55 | 2,70 | 3,70 | 4,70 | 6,70 | 8,70 | 19,0 |
В приведенных выше расчетных формулах:
tз - продолжительность работы фильтра до момента, когда качество фильтрата начнет ухудшаться, называемая продолжительностью защитного действия загрузки фильтра;
tн - продолжительность работы фильтра до момента, когда потеря напора в загрузке доходит до максимально возможной величины;
x - толщина слоя загрузки, м;
Hпр - предельно возможная потеря напора в фильтре, м;
H0 - начальная потеря напора в фильтре, м;
h/t - скорость прироста потери напора за фильтроцикл, м/ч;
i0 - средний начальный гидравлический уклон в загрузке фильтра (безразмерная величина) вычисляется делением начальной потери напора H0 (м) на толщину слоя загрузки (м);

Отношение  зависит не только от (гранулометрического состава загрузки, но и от направления движения воды через загрузку; оно будет больше 1 при движении воды сверху вниз и меньше 1 при движении воды снизу вверх.
зависит не только от (гранулометрического состава загрузки, но и от направления движения воды через загрузку; оно будет больше 1 при движении воды сверху вниз и меньше 1 при движении воды снизу вверх.
Значения параметров фильтрования при иных по сравнению с эталонными значениями скорости фильтрования и размеров зерен загрузки определяются расчетом по следующим формулам:




В этих формулах все величины, помеченные звездочками, относятся к эталонным значениям скорости фильтрования V* = 10 м/ч и крупности зерен d* = 1 мм.
Если опыт был проведен при скорости фильтрования и крупности зерен загрузки, отличающихся от эталонных, то прежде всего с помощью приведенных выше формул следует определить значения параметров  , i0, соответствующие эталонным значениям скорости фильтрования и крупности зерен.
, i0, соответствующие эталонным значениям скорости фильтрования и крупности зерен.
, i0, соответствующие эталонным значениям скорости фильтрования и крупности зерен.Оптимизация работы фильтровальных сооружений заключается в подборе условий, при которых:

где  - коэффициент запаса, принимаемый равным 1,2 - 1,3;
- коэффициент запаса, принимаемый равным 1,2 - 1,3;
tц - продолжительность работы фильтра до промывки, принимаемая в соответствии с требованиями СНиП.
Полученные значения параметров загрузки и скорости фильтрования проверяют опытом на лабораторных и полупроизводственных установках.
КОНТАКТНЫХ ОСВЕТЛИТЕЛЕЙ
54.1. Технологический анализ проводят для определения основных технологических параметров контактных осветлителей при осветлении и обесцвечивании воды.
Экспериментальная установка
54.2. Фильтровальная установка состоит из колонки диаметром 150 - 200 мм, высотой 3 м, оборудованной пьезометрами, а также системой для подачи и отвода воды (рис. 19).

основных технологических параметров контактных осветлителей
1 - подача речной воды; 2 - распределительный бачок;
3 - подача реагентов; 4 - воздухоотделитель; 5 - колонка
(модель контактного осветлителя); 6 - дырчатое дно;
7 - подача воды в колонку; 8 - пьезометры;
9 - пробоотборники; 10 - отвод фильтрата и промывной воды;
11 - подача промывной воды; 12 - перелив; 13 - насадки
для регулирования расхода
Колонку последовательно загружают гравием и песком следующих фракций:
1) гравий  , на высоту 10 см (нижний слой);
, на высоту 10 см (нижний слой);
, на высоту 10 см (нижний слой);2) гравий  , на высоту 10 см; песок
, на высоту 10 см; песок  , на высоту 200 см (эквивалентный диаметр зерен песка 0,9 - 1,3 мм).
, на высоту 200 см (эквивалентный диаметр зерен песка 0,9 - 1,3 мм).
, на высоту 10 см; песок , на высоту 200 см (эквивалентный диаметр зерен песка 0,9 - 1,3 мм).Приборы
54.3. Фотоэлектроколориметр или другие приборы, позволяющие определять мутность и цветность воды.
Мерные цилиндры или бачки емкостью 1 - 2 л.
Секундомер.
Реактивы
54.4. Сульфат алюминия, 1%-ный раствор по Al2(SO4)3 (приготовление см. стр. 247).
Проведение опыта
54.5. Определяют требуемую дозу коагулянта в соответствии с результатами определения по методике, приведенной на стр. 249. Промывают загрузку колонки восходящим потоком воды. В баке с исследуемой водой подбирают насадку для обеспечения скорости фильтрования в пределах 5 - 6 м/ч. Настраивают дозатор коагулянта на заданную дозу.
Производят фильтрование воды снизу вверх. В ходе опыта каждый час отбирают пробы исходной и профильтрованной воды. Измеряют объемным способом расходы воды и раствора коагулянта, по показаниям пьезометров записывают потерю напора в фильтрующей загрузке.
В пробах воды определяют мутность и цветность.
Если качество фильтрата ниже предъявляемых к нему требований, дозу коагулянта увеличивают. Проверяют также возможность снижения дозы коагулянта. Экспериментальные данные записывают в журнал наблюдений и оформляют в виде графиков (рис. 16 и 17).
Начиная с момента ухудшения качества фильтрата, его мутность и цветность определяют каждые 15 - 30 мин до предельно допустимого ухудшения качества фильтрата. Общее время работы контактного осветлителя до этого момента является временем защитного действия загрузки (tз). По рис. 17 определяют также время достижения предельной потери напора (tн).
Испытания проводят по три раза с двумя-тремя скоростями фильтрования, в пределах 5 - 6 м/ч в каждый характерный период года. По полученным результатам судят о целесообразности использования контактного осветления и об основных показателях работы контактных осветлителей при очистке исследуемой воды.
Общие положения
55.1. Исследования по хлорированию воды имеют целью определение дозы хлора, введение которой необходимо для окисления органических загрязнений и создания достаточной для обеззараживания воды концентрации остаточного хлора.
За необходимую дозу принимают наименьшую дозу хлора, введение которой в воду обеспечивает через 30 мин концентрацию остаточного свободного (не связанного в хлорамины) хлора 0,5 мг/л либо связанного в хлорамины хлора 1 мг/л.
Реактивы
55.2. Хлорная вода: готовят насыщением дистиллированной воды газообразным хлором или растворением хлорной извести до концентрации свободного хлора 100 мг/л. Концентрацию в растворе активного хлора проверяют по методике, описанной на стр. 226.
Ход определения
55.3. В 10 конических колб емкостью 250 мл с хорошо подогнанными пробками вливают пипеткой по 100 мл исследуемой воды. Затем вводят в первую колбу 0,5 мл хлорной воды, во вторую 1 мл, в третью - 1,5 мл и т.д. Колбы закрывают пробками, содержимое перемешивают и оставляют в покое на 30 мин, через 30 мин определяют содержание в воде свободного и активного хлора. По полученным данным строят график (рис. 20), где по оси абсцисс откладывают дозы введенного хлора в мг/л, а по оси ординат - количество остаточного свободного и активного хлора в мг/л.

I - моно- и дихлорамины; II - свободный хлор;
a - точка перелома
Необходимой дозой является количество введенного хлора, при котором концентрация остаточного свободного хлора в воде равна 0,5 мг/л (после точки перелома a, соответствующей окончанию окисления хлораминов) либо остаточного хлораминного хлора (до точки перелома a) - 1,0 мг/л.
56.1. Под стабильностью воды понимают ее свойство не растворять карбонат кальция и не выделять его из раствора.
Стабильность воды обусловлена равновесным состоянием в растворе углекислых соединений

и характеризуется тем, что концентрация свободной двуокиси углерода (CO2) соответствует ее равновесной концентрации, необходимой для поддержания в растворе содержащихся в воде солей угольной кислоты.
Стабильность воды определяется двумя методами:
1) карбонатными испытаниями (экспериментальный метод);
2) вычислением по данным анализов воды индекса насыщения раствора карбонатом кальция (приближенный расчетный метод).
Карбонатными испытаниями учитывается влияние содержащихся в воде соединений, сорбирующихся на поверхности раздела фаз и задерживающих кристаллизацию или растворение карбоната кальция.
При вычислении индекса насыщения раствора карбонатом кальция влияние этого фактора не учитывается.
Метод карбонатных испытаний
Аппаратура
56.2. Встряхивающий аппарат с качающейся платформой для встряхивания сосудов с исследуемой водой.
Бюретка.
pH-метр.
Приспособление для отбора воды из сосуда после встряхивания пробы воды с порошкообразным карбонатом кальция (рис. 21). Приспособление состоит из сосуда с резиновой пробкой 2, по размеру соответствующей горлышку сосуда, через которую пропущены хлоркальциевая трубка 3, заполненная натронной известью, и стеклянная трубка 4, не доходящая на 3 - 4 см до дна сосуда. На верхний конец стеклянной трубки надевается резиновый шланг длиной 75 - 80 см, другой конец шланга соединен со стеклянной трубкой 4, пропущенной через резиновую пробку 5.

1 - сосуд; 2 и 5 - резиновые пробки; 3 - хлоркальциевая
трубка; 4 - стеклянные трубки; 6 - воронка
со стеклянным фильтром
Эта пробка вставлена в воронку 6 с фильтром из пористой стеклянной пластинки N 2 (размер пор 40 - 50 мк).
Реактивы
56.3. Соляная или серная кислота, 0,1 н. раствор (приготовление раствора и определение поправочного коэффициента см. стр. 42).
Метиловый оранжевый, 0,1%-иый водный раствор. Приготовление см. стр. 36.
Карбонат кальция, порошкообразный: 250 г кристаллического хлорида кальция (CaCl2·6H2O) растворяют в 1 л дистиллированной воды; в другом сосуде растворяют 175 г карбоната аммония (NH4)2CO3 в 1,5 л дистиллированной воды. Сливают оба раствора в цилиндрический сосуд емкостью 3 - 5 л и непрерывно перемешивают в течение 15 - 20 мин. Образовавшемуся осадку карбоната кальция дают осесть в течение 2 - 3 ч. Воду над осадком сливают, оставшийся на дне осадок переносят частями в воронку Бюхнера с уложенным на дно кружком фильтровальной бумаги и отмывают дистиллированной водой до отсутствия хлоридов в промывной воде. Промытый осадок высушивают при 110 °C и прокаливают при 300 - 350 °C в течение 3 - 4 ч.
Ход определения
56.4. Пробу воды объемом не менее 2 л доставляют в лабораторию для исследования в стеклянной бутыли, заполненной до пробки. Определение следует производить по возможности немедленно, но не позднее, чем через 2 ч после отбора пробы. Если вода содержит взвешенные вещества, пробу отстаивают в течение 1 - 2 ч. Отбирают 100 мл исследуемой воды в коническую колбу и титруют 0,1 н. соляной (или серной) кислотой для определения щелочности воды.
Затем в прочный стеклянный сосуд емкостью 400 - 500 мл всыпают 30 г порошкообразного карбоната кальция, заполняют сосуд до пробки исследуемой водой, плотно закрывают резиновой пробкой, устанавливают в горизонтальном положении на платформу встряхивающего аппарата и в течение 3 ч встряхивают при 100 - 150 качаниях платформы в минуту.
Желательно обеспечить такие условия проведения опыта, при которых выдерживалась бы температура исследуемой воды, соответствующая ее температуре при отборе пробы.
При необходимости оценивать стабильность воды при нагревании ее до заданной температуры поступают следующим образом. Перед испытаниями воду нагревают до нужной температуры в заполненной доверху колбе, закрытой пробкой с небольшим отверстием. Затем подогретую воду переливают в термос емкостью 0,5 л или сосуд, покрытый термоизоляцией. Предварительно термос или сосуд нагревают до температуры исследуемой воды двух-трехкратным заполнением нагретой дистиллированной водой.
По окончании встряхивания снимают сосуд со встряхивающего аппарата, быстро заменяют пробку приспособлением для отбора воды (рис. 21) и оставляют в покое на 1 ч для осаждения порошка карбоната кальция.
После осветления воды сифоном отсасывают воду через нижний конец воронки со стеклянным фильтром, сбрасывают первые 50 мл воды и отбирают в коническую колбу 100 мл фильтрованной воды. Эту пробу титруют 0,1 н. раствором HCl (или 0,1 н. раствором H2SO4) с индикатором метиловым оранжевым.
Расчет. Вычисляют щелочность воды (стр. 42) и рассчитывают показатель стабильности воды (мг-экв/л), определенный по методу карбонатных испытаний (C), по формуле
C = Щ1 - Щ2,
где Щ1 - щелочность исходной воды, мг-экв/л;
Щ2 - щелочность воды после встряхивания с порошкообразным карбонатом кальция, мг-экв/л.
Вычисление индекса насыщения карбонатом кальция
56.5. Вычисление производят на основе следующих данных: 1) температура воды (в момент отбора пробы или после нагревания до заданной температуры в случае, если необходимо оценить стабильность воды после ее нагрева); 2) содержание кальция; 3) щелочность; 4) общее содержание солей (приближенно принимать равным сухому остатку); 5) pH.
Расчет. Величину pH равновесного насыщения воды карбонатом кальция вычисляют по формуле
pHs = f1(t) - f2(Ca2+) - f3(Щ) + f4(P),
где pHs - величина pH равновесного насыщения воды карбонатом кальция;
f1(t), f2(Ca2+), f3(Щ), f4(P) - величины, зависящие соответственно от температуры воды, содержания в ней кальция, щелочности и общего содержания солей. Определяют эти величины по соответствующим шкалам номограммы на рис. 22.

при вычислении индекса насыщения воды карбонатом кальция
Индекс насыщения воды карбонатом кальция (J) вычисляют по формуле
J = pH0 - pHs,
где pH0 - измеренное с помощью pH-метра значение pH исследуемой воды.
Примечание. Величину pH0 обычно измеряют при температуре 18 - 20 °C. Если индекс насыщения нужно рассчитать для нагретой воды, то в измеренную при 18 - 20 °C величину pHизм нужно внести поправку в соответствии с формулой 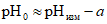 , где pHизм - величина pH воды, измеренная при 18 - 20 °C;
, где pHизм - величина pH воды, измеренная при 18 - 20 °C;
, где pHизм - величина pH воды, измеренная при 18 - 20 °C;a - температурная поправка, которую находят по табл. 15.
Таблица 15
после ее нагрева
pH при температуре 18 - 20 °C pHизм | Температура нагретой воды в °C | Температурная поправка (a) при щелочности воды, мг-экв/л | |||||
0,5 | 1 | 2 | 4 | 8 | |||
<= | 8,0 | 50 | 0,1 | 0,1 | 0,1 | 0,1 | 0,1 |
8,2 | 0,2 | 0,15 | 0,15 | 0,15 | 0,1 | ||
8,4 | 0,3 | 0,2 | 0,2 | 0,15 | 0,15 | ||
<= | 7,6 | 60 | 0,1 | 0,1 | 0,1 | 0,1 | 0,1 |
7,8 | 0,15 | 0,15 | 0,1 | 0,1 | 0,1 | ||
8,0 | 0,3 | 0,2 | 0,15 | 0,15 | 0,1 | ||
8,2 | 0,4 | 0,3 | 0,2 | 0,2 | 0,15 | ||
8,4 | 0,5 | 0,4 | 0,3 | 0,25 | 0,2 | ||
Оценка результатов
56.6. Вода считается стабильной, если показатель стабильности C или индекс насыщения J равны нулю. При положительных значениях C или J из раствора может выделяться карбонат кальция, чем обусловлена способность воды создавать на омываемой поверхности труб защитную против коррозии карбонатную пленку.
При отрицательных значениях C или J вода содержит агрессивную углекислоту, в связи с чем стимулируется коррозия стальных и чугунных труб, а в определенных условиях и бетона <*>.
--------------------------------
<*> Для оценки агрессивного воздействия воды на бетон железобетонных конструкций надлежит руководствоваться "Указаниями по проектированию антикоррозионной защиты строительных конструкций", СН 262-67.
57.1. Целью исследования является получение данных для выбора метода обезжелезивания и установления необходимых доз реагентов.
Обезжелезивание подземных вод
57.2. Пробное обезжелезивание подземных вод проводят с целью выбора одного из двух следующих методов: 1) фильтрования с упрощенной аэрацией; 2) аэрации в сочетании с обработкой хлором или перманганатом калия.
В первую очередь определяют возможность применения фильтрования с упрощенной аэрацией.
Обезжелезивание воды фильтрованием с упрощенной аэрацией
Реактивы
57.3. Кислота соляная, уд. плотность 1,12.
Реактивы и посуда для определения железа (см. стр. 95).
Ход определения
57.4. Определение производят непосредственно на месте отбора проб, перевозка проб на расстояние не допускается. Исследуемую воду медленно со свободным изливом наливают в стеклянный сосуд с открытой поверхностью емкостью 10 - 12 л.
После заполнения сосуда из среднего слоя исследуемой воды через 0,25; 0,5; 1,0; 2,0; 3,0 ч пипеткой отбирают в две колбы пробы объемом по 50 мл.
Для консервирования проб в колбы предварительно вносят 1 - 2 мл соляной кислоты уд. плотностью 1,12. В одной колбе определяют содержание окисного железа, в другой - общего (см. стр. 94). По разности между концентрациями общего и окисного железа определяют содержание закисного железа в пробе.
По полученным данным рассчитывают параметр  - показатель скорости окисления:
- показатель скорости окисления:

где Feобщ - содержание общего железа в исходной воде, мг/л;
Feзак - содержание закисного железа в данный момент времени, мг/л;
t - время нахождения воды в сосуде перед отбором проб (0,25; 0,5; 1,0; и т.д.), ч.
Принимают среднее арифметическое значение  из пяти определений.
из пяти определений.
Метод фильтрования с упрощенной аэрацией может быть рекомендован при значении  более 0,06 <*>.
более 0,06 <*>.
--------------------------------
<*> НИИ КВ и ОВ АКХ "Технические условия на проектирование и эксплуатацию установок по обезжелезиванию воды фильтрованием", 1972.
По полученному значению  , исходя из данных табл. 16, определяют также продолжительность "зарядки" загрузки фильтра.
, исходя из данных табл. 16, определяют также продолжительность "зарядки" загрузки фильтра.
Примечание. Продолжительностью "зарядки" загрузки фильтра называется время, необходимое для образования на зернах загрузки каталитической пленки, позволяющей получить качество фильтрата с содержанием железа менее 0,3 мг/л.
Показатель скорости окисления | Продолжительность зарядки (загрузки), ч | Показатель скорости окисления | Продолжительность зарядки (загрузки), ч |
0,6 | До 50 | 0,14 | До 200 |
0,3 | " 70 | 0,1 | " 240 |
0,22 | " 120 |
Обезжелезивание воды аэрацией в сочетании
с обработкой хлором или перманганатом калия
Аппаратура
57.5. Песчаный фильтр (стеклянная трубка диаметром 40 - 50 мм, загруженная песком крупностью 0,5 - 1,2 мм на высоту 500 мм).
Стеклянные сосуды объемом 3 л.
Реактивы
57.6. Хлорная вода (приготовление см. стр. 248).
Перманганат калия - 0,1%-ный водный раствор.
Кроме того, необходимы аппаратура и реактивы для определения общего и окисного железа (см. стр. 94).
Ход определения
57.7. Определение дозы хлора. Определение производят непосредственно на месте отбора проб. Испытуемую воду наливают в пять сосудов емкостью по 3 л.
В первый сосуд вносят хлорную воду в количестве, соответствующем 0,5 мг/л активного хлора. Воду, после введения хлора, перемешивают стеклянной палочкой в течение 1 мин. Через 5 - 10 мин воду фильтруют через песчаный фильтр, первые порции фильтрата отбрасывают и затем определяют содержание общего железа в фильтрате. После фильтрования всей воды песок в фильтре промывают током воды снизу вверх. Затем хлорную воду вносят поочередно во второй, третий, четвертый и пятый сосуды в количествах, обеспечивающих соответственно 1 мг/л, 3 мг/л, 5 мг/л и 8 мг/л. После чего воду фильтруют через песчаный фильтр, как указано выше. В качестве рекомендуемой принимается наименьшая доза хлора, при которой происходит практически полное обезжелезивание воды (содержание общего железа в фильтрате не должно превышать 0,3 мг/л).
57.8. Определение дозы перманганата калия. Определение дозы перманганата калия производят аналогично определению дозы хлора. В пробы воды добавляют соответствующие количества 0,1%-ного раствора KMnO4.
В качестве рекомендуемой принимается наименьшая доза перманганата калия, которая обеспечивает остаточные концентрации общего железа в воде после фильтрования через песок не более 0,3 мг/л (при использовании воды для питьевых целей) и введенного с перманганатом калия марганца не более 0,1 мг/л (определение марганца см. стр. 121).
Обезжелезивание поверхностных вод
57.9. Обезжелезивание поверхностных вод производят одновременно с их осветлением и обесцвечиванием.
Дозу коагулянта для обезжелезивания принимают равной оптимальной дозе коагулянта, определенной из условий осветления и обесцвечивания исследуемой воды (см. стр. 247).
Если указанная доза не обеспечивает нужную степень обезжелезивания (содержание общего железа в фильтрате не снижается до 0,3 мг/л), дополнительно к коагулянту вводят хлор, перманганат калия или известь.
Определение дозы хлора и перманганата калия
57.10. Для определения дозы хлора в пять стеклянных сосудов объемом по 3 л отбирают пробы испытуемой воды. В каждый сосуд вводят дозу хлора из расчета 0,5 мг/л, 1 мг/л, 3 мг/л и т.д. и оптимальную дозу коагулянта. После введения хлора и коагулянта содержимое сосуда тщательно перемешивают стеклянной палочкой и оставляют в покое на 1 ч. Аналогично проводят определение с перманганатом калия.
Дальнейший ход определения такой же, как при определении дозы хлора и перманганата калия. В качестве рекомендуемой принимают наименьшую дозу хлора или перманганата калия, которые обеспечивают практически полное обезжелезивание воды.
Определение дозы извести
57.11. Для определения дозы извести в стеклянные сосуды объемом по 3 л отбирают пробы испытуемой воды. В каждый сосуд вводят известь и оптимальную дозу коагулянта. Дозу извести (мг/л) принимают из расчета: в первый сосуд - в соответствии с формулой

где ДCaO - доза извести, мг/л;
CO2 - количество свободной углекислоты, мг/л;
Дк - доза коагулянта (сульфата алюминия в расчете на Al2(SO4)3), мг/л.
Во второй сосуд вводят 50% и в третий - 150% рассчитанной дозы.
Содержимое сосудов тщательно перемешивают стеклянной палочкой и оставляют в покое на 1 ч. Дальнейший ход определения аналогичен описанному на стр. 248.
В качестве рекомендуемой принимают минимальную дозу, при которой обеспечивается требуемая степень обезжелезивания.
"Настоящий стандарт устанавливает методы определения общих физических свойств хозяйственно-питьевой воды: запаха, вкуса и привкуса, температуры, прозрачности, мутности, взвешенных веществ и цветности.
5 4 1 7 8 3 0 9".